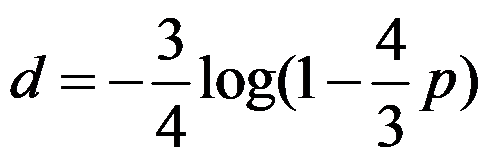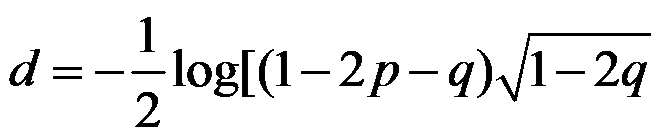Approche génomique
Introduction à l'évolution moléculaire
Aujourd’hui l’évolution moléculaire est utilisée non seulement
par les spécialistes de la phylogénie mais aussi par de nombreux biologistes
désirant mieux analyser leurs séquences, comprendre l’évolution de leur
fonction, analyser l’histoire des duplications etc….
Pour cela il faut entre autre connaître :
• les différents modèles évolutifs qui ont été proposés
• les différentes méthodes de reconstruction d’arbres qui
ont été développées
• apprendre à analyser les arbres obtenus
La reconstruction phylogénétique :
Le pourcentage de similarité entre deux séquences est
considéré comme reflétant la distance évolutive existant entre ces deux
séquences. Les différences observées sont dues à l’accumulation de mutations au
cours du temps. Les mutations prises en compte sont les substitutions et les
insertions/délétions (indels).
Donc à partir d'un alignement multiple et donc de la comparaison
des états de caractères (un caractére étant
ici une position alignée), il va falloir reconstruire l'arbre et
interpréter les ressemblances.
Deux écoles :
- Les phénéticiens adeptes de
la « taxonomie numérique ». Les liens entre les taxons ne peuvent
être fondés que sur la base d’une similitude globale exprimée à partir de
matrices de calcul de distances. Dans le cas des séquences, à partir d’un
alignement multiple, on calculera les distances entre les séquences prises deux
à deux en prenant en compte toutes les positions alignées sans indels.
L’analyse phénétique se fonde
sur l’analyse du plus grand nombre de caractères.
- Les cladistes préfèrent
élaborer des phylogénies à partir d’un
ensemble préalablement choisi de
caractères car lorsque l'on observe une similitude elle peut
soit être réellement une homologie, similitude
héritée d'un ancêtre commun, ou bien une homoplasie
similitude non héritée d'un ancêtre commun, ce qui
inclut la convergence - apparition indépendante dans deux
espèces du même état de caractère (par
exemple même substitution) - et la réversion - apparition
d'un état de caractère ayant la forme ancestrale.
Problème : Trouver l'arbre vrai
Il y a un seul arbre vrai , l'arbre évolutif, mais comment le
trouver parmi tous les scenaris et donc parmi tous les arbres possibles. En
effet, pour 10 séquences on a 2 027 024 arbres non
enracinés possibles et un seul vrai que l'on considère
comme le plus parcimonieux. Quand le nombre de séquences devient
supérieur à 10, on ne peut pas explorer tous les arbres
possibles pour en déduire l'arbre vrai. On fait donc appel
à des heuristiques.
Quatre familles principales de méthodes :
•
Parcimonie : à partir d’un ensemble de caractères choisis.
• Méthodes
de distance : à partir de distances établies sur un ensemble de caractères.
• Méthodes
du maximum de vraisemblance : à partir des probabilités de l’apparition des
transformation d’un état de caractères en un autre.
• Approche bayésienne
Pour les méthodes de distance et de maximum de vraisemblance, il va falloir choisir le modèle évolutif.
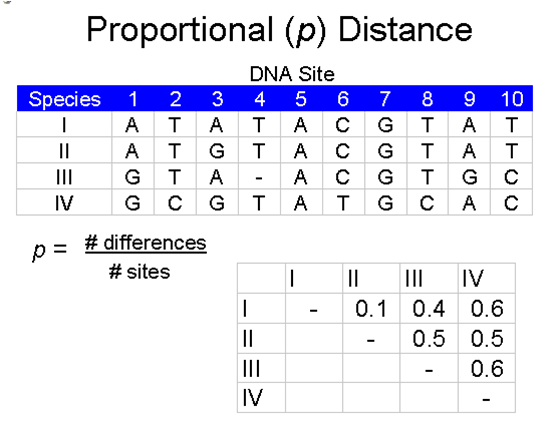
Calcul des distances entre deux séquences d'acides nucléiques (à partir des séquences alignées par alignement multiple)
La distance d séparant deux séquences est définie comme le nombre moyen
de substitution par site qui s’est produit depuis que ces deux séquences ont
divergé de leur ancêtre commun. La distance la plus simple est la distance observée appelée p-distance. On compte le nombre s de
substitutions observées entre deux séquences alignées que l’on rapporte au
nombre de sites homologues n alignés, donc p =s/n.
exemple :
 |
facile à
calculer mais quand les séquences ne sont pas proches (issues d’organismes
distants dans l’évolution), elle sous-estime les distances évolutives.
Cause :
l’existence de substitutions multiples. Phénomène plus critique pour les
séquences d’acides nucléiques car possèdent un alphabet plus pauvre que les
séquences protéiques : quatre lettres au lieu de 20.
|
Exemples de substitutions multiples conduisant à une sous-estimation des distances :
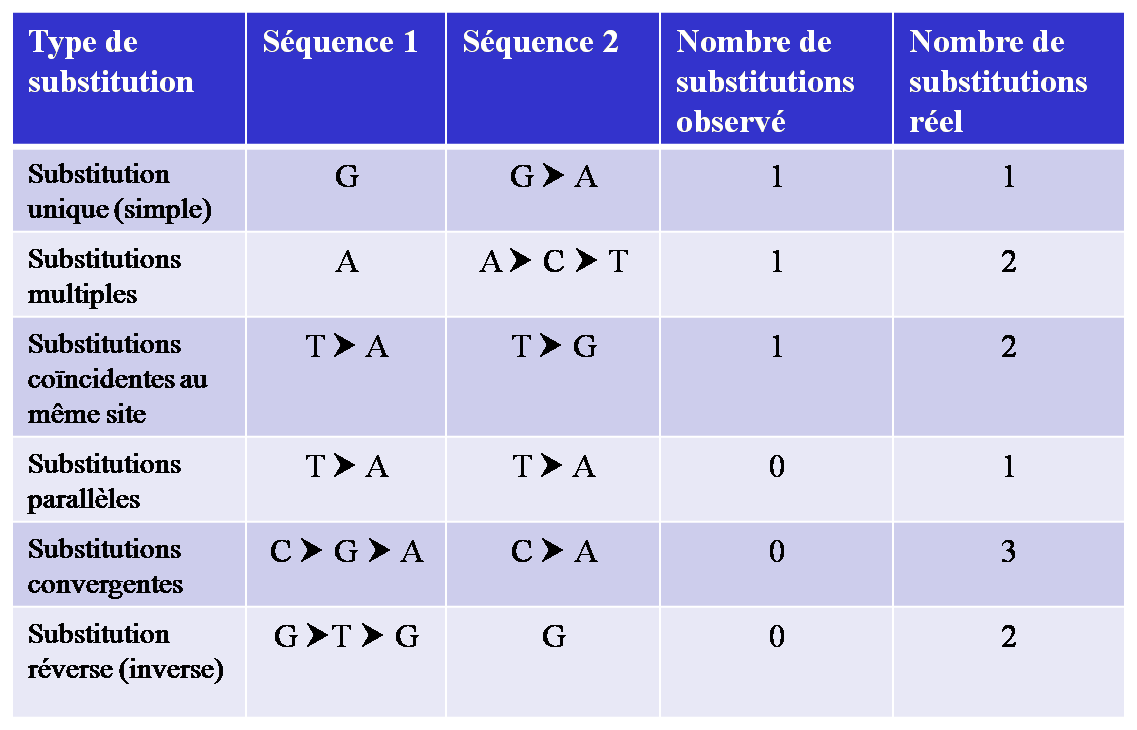 |
Séquence1
GAAAAG
Séquence2
ATGAAG
|
Pour
tenter de corriger le biais du aux mutations
multiples, des hypothèses sont faites sur la façon dont
les bases se sont
substituées à un locus donné -> construction
d'un modèle évolutif. Il existe plusieurs modèle
pour rendre compte de l'évolution des séquences.
Le plus simple, le modèle de Jukes et Cantor mais vision simplificatrice de l'évolution des séquences : i) tous
les sites évoluent indépendamment et selon le même
processus et ii) toutes les substitutions sont équiprobables. La
distance de Jukes et Cantor est donnée par la formule suivante
ou p est la p-distance.
Autre distance : distance de Kimura à 2 paramètres, vision
un peu moins simplificatrice : i) tous les sites évoluent indépendamment et selon le même
processus et ii) les substitutions se produisent suivant deux taux
distincts, l’un pour les transitions, l’autre pour les transversions, les
transitions étant plus fréquentes
(transition
= A<->G ou T<->C). La distance de Kimura deux
paramètres est donnée par la formule suivante où p
et q sont respectivement les fréquences observées des
transitions et des tranversions.
Pour les autres distances voir Perrière et Brochier-Armanet (2010) Concepts et méthodes en phylogénie moléculaire.
Calcul des distances entre deux séquences protéiques (à partir des séquences alignées par alignement multiple)
Séquences protéiques fréquemment utilisées en phylogénie
moléculaire car plus appropriées quand les analyses comportent des séquences
issues de lignées séparées par de grandes distances évolutives ou quand les
séquences évoluent rapidement (au niveau ADN perte du signal phylogénétique car
les sites sont dits saturés, i.e.,
ont subi de
nombreuses substitutions multiples). Egalement plusieurs modèles
pour estimer la distance entre deux séquences protéiques.
Le modèle de Poisson :
P Première
estimation meilleure que la p-distance repose sur le concept de distribution de
Poisson. Hypothèses
:i) tous les sites évoluent indépendamment et suivant un même processus et ii) toutes les substitutions sont équiprobables, iii) le taux de réversion est négligeable.
La correction de poisson est donnée par d = -Log (1-p) avec p valeur de la p-distance
Cependant vision simplificatrice de l'évolution car on sait que :
- taux de substitutions plus ou moins élevé en
fonction de l’importance fonctionnelle du site
- présence aussi de substitution parallèle et de
réversion donc on va sous-estimer la distance entre deux séquences
- ne peut être utilisée que si séquences
globalement peu divergentes
Donc d'autres modèles ont été développés qui ne seront pas détaillés :
- Le modèle PAM
- Le modèle JTT
- Le modèle WAG
- Le modèle LG
Les modèles les plus performants sont ceux bâtis sur
le plus grand nombre de données (car estimation des taux de substitutions pour
tous les modèles). Les modèles WAG et LG sont supérieurs aux modèles PAM et JTT. Cependant, si
les distances évolutives sont faibles, on peut utiliser le modèle de Poisson ou de
Kimura car aussi bons résultats. Donc si
même résultat avec deux modèles, utiliser le plus simple car plus rapide.
Choix d'un modèle évolutif
Des méthodes permettant de tester l’adéquation du modèle
aux données existent mais souvent le choix du modèle est du fait de
l’utilisateur et de ses connaissances.
Quelques
règles simples :
construction d’une phylogénie à partir de gènes protéiques :
• séquences très distantes dans l’évolution : utilisation
des séquences protéiques.
• séquences proches dans l’évolution : utilisation des
séquences acides nucléiques voir
travailler uniquement sur les positions synonymes.
Utilisation
de séquences nucléiques : grand nombre de modèles
• critère important : le degré de divergence entre les
séquences.
• pas toujours pertinent d’utiliser les modèles avec
beaucoup de paramètres :
- si les séquences sont courtes ou trop similaires les
estimations des paramètres sont mauvaises.
- modèle arrivant à saturation plus rapidement donc si
séquences très divergentes, fréquemment impossible de calculer les distances.
• donc si même résultat avec deux modèles, utiliser le plus
simple car la variance de la distance augmente avec le nombre de paramètres.
• application de la correction Gamma que si nombre de sites
utilisés important car nécessite
d’estimer un paramètre supplémentaire (la forme de la distribution).
Choix d'une méthode de reconstruction phylogénétique :
Méthode de parcimonie : cherche à minimiser le nombre de changements. Plus adéquate
pour des données de présence/absence. Pas conseillée pour la reconstruction
phylogénétique à partir des séquences.
Méthode de distance : la Neighbor Joining (NJ) : elle constitue une approximation du minimum d’évolution (critère
d’optimisation). Le principe général du minimum d’évolution est le suivant : Examine toutes
les topologies, calcule la somme de la longueur des branches de chacune
d’entre-elles et retient celle qui minimise la somme des longueur des branches
(arbre de longueur minimum). Des variantes avec pondérations ont également été développées
comme la BioNJ (Gascuel, 1997). La BioNJ apporte de améliorations évidentes surtout
quand les séquences sont fortement divergentes et/ou quand elles présentent des
vitesses d’évolution différentes.
Conclusion :
- Méthode performante
car bon équilibre entre rapidité et efficacité. Elle peut être appliquée sur
des très grands jeux de données. Robuste car ne dépend pas de l’ordre des
séquences.
- ne fait pas
l’hypothèse de l’horloge moléculaire (n’impose pas aux distances estimées
d’être ultra-métriques).
- Souvent utilisée
pour chercher des arbres qui vont servir de point de départ pour des méthodes
plus coûteuses en temps calcul comme la méthode du maximum de vraisemblance
(cf. ce chapitre du cours).
- Elle peut être
appliquée sur n’importe quel type de distances évolutives.
- Peut conduire à des
distances négatives notamment pour les branches terminales mais l’application
de la contrainte de non-négativité permet de s’affranchir du problème.
- Problème pouvant
être rencontré quand deux paires de voisins différentes donnent des arbres
minimums de même longueur. Dans ce cas, tirage au hasard d’une solution.
Situation pas fréquemment rencontrée.
- Ne donne pas
d’informations sur les états de caractères de l’ancêtre commun.
Méthode du maximum de vraisemblance : deux méthodes PhyML et Tree-Puzzle :
On a un
grand nombre de scénarios évolutifs possibles. Cependant certains d’entre eux
sont plus susceptibles que d’autres de produire les séquences actuelles.
Le but des
méthodes de maximum de vraisemblance est d’identifier ces scénarios,
c’est-à-dire de trouver les valeurs des paramètres qui maximisent la
probabilité d’observer les séquences actuelles.
Conclusions
C’est la méthode la mieux justifiée
au plan théorique.
Des expériences de simulation de
séquences ont montré que cette méthode est supérieure aux autres dans la
plupart des cas.
Mais c’est une méthode très lourde
en calculs.
Il est presque toujours impossible
d’évaluer tous les arbres possibles car ils sont trop nombreux. Une exploration partielle de l’ensemble des
arbres est réalisée en utilisant des méthodes de réarrangements locales ou
globales.
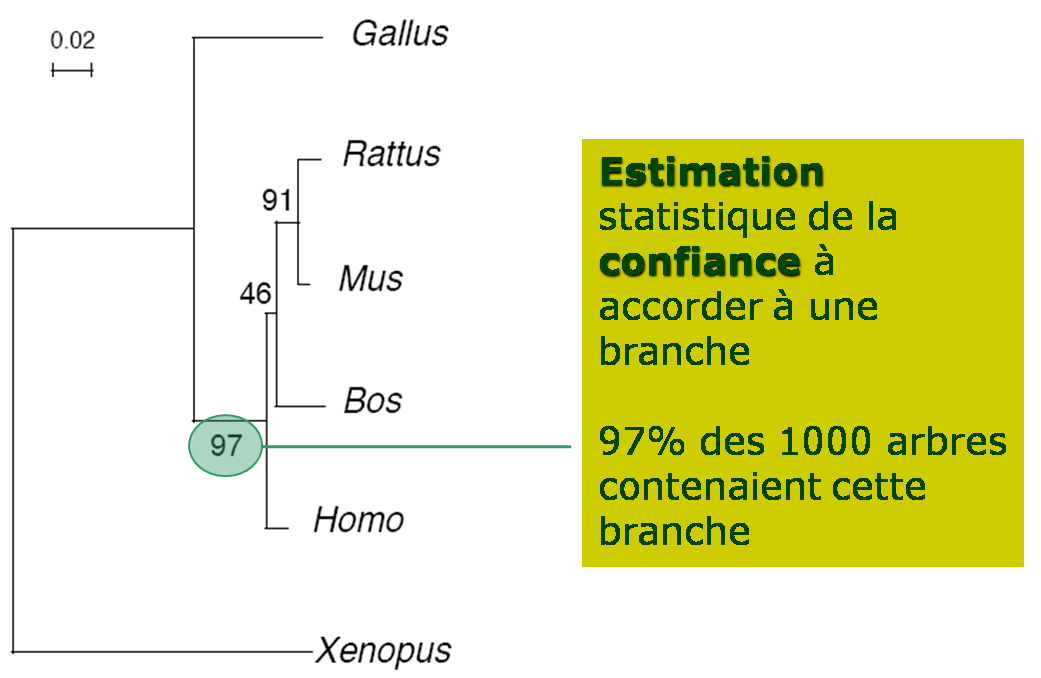
Evaluation de la fiabilité de la topologie obtenue : le bootstrap
La
forme de l'arbre est déterminée par la liste de ces
branches internes. Evaluer la fiabilité de l'arbre =
évaluer celle de chacune de ces branches internes.
Ceci est réalisé par la procédure du bootstrap.
On
part de notre alignement multiple et on reconstruit un alignement de
même longueur par tirage aléatoire avec remise des
positions de l'alignements.
Alignement de départ : chaque position alignée = une couleur (n positions)

On effectue un tirage
avec remise de n positions parmi n positions. On obtient un nouvel
alignement de même longueur où certaines positions peuvent
être présentes plusieurs fois (bleu clair par exemple) ou
absente (noire par exemple).
Nouvel alignement :
En utilisant ce nouvel alignement on reconstruit un arbre.
On réitère : tirage avec remise pour obtenir l'alignement
multiple et construction de l'arbre un grand nombre de fois (500
à 1000).
Pour chaque arbre reconstruit on compte le nombre de fois
que l’on observe la branche interne. Le soutien de la branche est exprimé en
pourcentage de réplications. Si la branche est observée dans tous les arbres,
la valeur du bootstrap est égale à 100.
exemple :
Interprétation des valeurs de bootstrap :
Problème très discuté! De manière
générale, une faible valeur de bootstrap indique que la quantité d’information supportant la
bipartition induite par une branche interne est faible.
Quel seuil ?
Si on applique les critères standards utilisés
en statistique, il ne faudrait considérer comme valide que les branches ayant
un support de bootstrap ≥ 95% (sinon la branche n’existe pas). Des travaux
ont montré que ce seuil était trop élevé, notamment ceux de Hillis et Bull
(1993, Syst. Biol., 42,
182-92) qui à l’aide de simulations ont montré que des supports de 70%
pouvaient correspondre à des groupements significatifs.
Cependant
résultat pas généralisable à toutes les analyses.