Création d'un HMM pour prédire les promoteurs de type sigmaA de Bacillus subtilis
Installation du programme
Télécharger l'archive compressée dans le
répertoire Software :
- pour le système d'exploitation Linux Debian DEBIAN
- pour le système d'exploitation Linux Fedora FEDORA
Une fois décompressée et désarchivée (tar -xvf SHOW.tar.gz), un répertoire show-debian-etudiant est
créé dans lequel vous trouverez les différents
sous-répertoires nécessaires au programme, deux fichier sigma_em et sigma_vit qui vous seront utiles par la suite ainsi qu'un sous-répertoire seqdna contenant un ensemble de séquences de régions promotrices de Bacillus subtilis qui vous permettront d'apprendre les paramètres de votre modèle HMM et de le tester.
Pour tester si cela marche :
Se mettre dans le répertoire show-debian-etudiant et lancer la commande suivante :
bin/show_emfit
Si vous obtenez une réponse commençant par Usage :.... C'est que c'est bon
Si vous avez l'erreur suivante :
"error while loading shared libraries libgsl.so.0",
c'est que le chemin pour utiliser cette librairie n'est pas
reconnu. Il faut donc expliciter ce chemin. La librairie se trouve dans
show-debian-etudiant/lib.
Faire pwd pour avoir le chemin complet de votre directory show-debian-etudiant
Ensuite sur le terminal taper les deux commandes suivantes:
LD_LIBRARY_PATH= le résultat de pwd/lib
export LD_LIBRARY_PATH
Refaire bin/show_emfit. Maintenant cela doit être bon
Les séquences de travail
Comme dit ci-dessus, elles se trouve dans le
sous-répertoire seqdna dans show-debian-etudiant. Chaque séquence se trouve dans un
fichier individuel en format Fasta ayant l'extension .dna. Sur la
première ligne à la suite du nom de la séquence
sont indiquées les positions connues des régions -35 et
-10 des promoteurs sigma A.
Création du modèle HMM
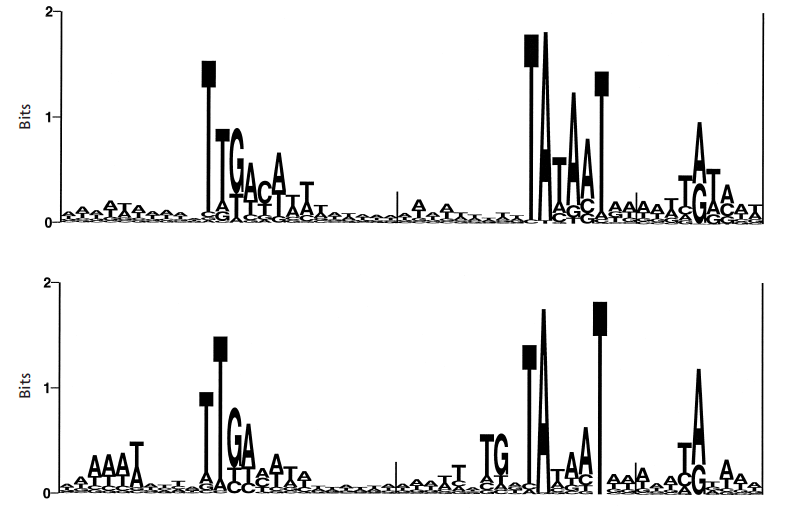
En vous basant sur les logos ci-dessous établir le
schéma de votre HMM où chaque position de la
séquence correspondra à un état. Deux logos
différents ont été établis pour
représenter les régions -35 et -10 ainsi que le +1 de
transcription pour une région promotrice de type sigma A normal
(le premier) ou une région promotrice étendue (le
second). Chaque logo a été obtenu en combinant 3 logos
obtenus indépendamment (petite barre verticale), un pour la
région -35, un pour la région -10 et un pour la
région +1. Un seul HMM sera établi pour tenter
d'identifier simultanément ces deux types de régions
promotrices dans les séquences de Bacillus subtilis.
Pour la modélisation, il faudra déterminer la taille de
chacune de vos régions conservées ainsi que la distance
entre ces régions.
La distance entre la boîte -35 et -10 peut varier de 16 à
22 nucléotides et la distance entre la boîte -10 et le +1
de transcription peut varier de 4 à 10 nucléotides.
Vous allez utiliser SHOW développé à l'INRA de
Jouy en Josas pour construire votre HMM. Pour cela il faut prendre
connaissance de sa syntaxe. Vous avez à votre disposition la
documentation fournie avec le programme ainsi qu'un extrait commenté de cette syntaxe.
Modélisation du HMM et implémentation dans la syntaxe de SHOW
Pour vous faciliter le travail un template pour construire le modèle
vous ai fourni. Il faudra le compléter. Ce template est disponible ici.
Une fois votre fichier modèle construit, il va falloir réaliser
l'estimation des paramètres du modèle (probabilités d'émission et de
transition) en utilisant les séquences qui vous ont été fournies. Pour
cela, en plus du fichiers modèle, deux autres fichiers sont requis
Préparation des fichiers pour SHOW :
- Fichier séquences :
Les exécutables de SHOW peuvent travailler soit sur une seule
séquence, soit sur un ensemble de séquences. Les séquences à analyser
sont référencer dans le fichier –seq<file>. Ce fichier doit suivre la structure suivante :
seq_identifier: sigma.dna
seq_type: dna
seq_files:
aadk.dna
abr.dna
...
seq_identifier fait référence à un identifiant choisi pour
la (les) séquence(s). Il doit être le même que celui donné dans le
fichier modèle (-model) pour le mot clef seq au niveau de la description des observations.
seq_type correspond à la nature des séquences (pour le moment seulement adn)
seq_files correspond aux noms des fichiers contenant les séquences à analyser
Pour créer ce fichier dans le répertoire seqdna contenant vos fichiers séquences, faire :
ls *.dna sigma_seq.txt (créera un fichier appelé sigma_seq.txt et contenant la lite des noms de vos fichiers)
Ajouter au début de ce fichier, les trois lignes seq_identifier, seq_type et seq_files
- Fichier contenant les informations pour initialiser et faire tourner l’algorithme :
Fichier –em<file>
Ce fichier sigma_em vous a été fourni dans l'archive. Il contient les lignes suivantes :
Les deux variables suivantes contiennent la taille des segments utilisée
pour la mémoire sauvant les approximations lors de l’étape
forward-backward de l’algorithme
estep-segment: 2000
estep_overlap: 100
nb_sel, niter_sel, eps_sel ne sont requises que si pobs vaut random
nb_sel: 3
niter_sel: 100
eps_sel: 0.01
niter: 1000
epsi: 0.001
niter et epsi: critère d’arrêt.
L’algorithme stop quand l’augmentation de la vraisemblance
entre deux itérations est plus petite que la valeur de epsi ou quand le nombre maximal d’itérations défini par niter est atteint.
nb_sel correspond au nombre de point de départ aléatoire de l’algorithme EM
niter_sel se rapporte au nombre maximal d’itérations réalisé par point de départ
eps_selcorrespond au critère d’arrêt de l’algorithme pour chaque point de départ
Etape d'estimation des paramètres :
L'estimation des paramètres du modèle (probabilités d'émission et de
transition) se fait en utilisant l'algorithme de Baum-Welch implémenté
dans SHOW. Pour cela il va falloir lancer la commande suivante en se
placant dans le répertoire contenant les fichiers séquences et votre
fichier sigma_seq.txt donc dans seqdna. :
../bin/show_emfit -model ../sigma_model -em ../sigma_em -seq sigma_seq.txt > /dev/null
La commande > /dev/null va rediriger les warnings qui s'affichent
à l'écran lorsque le programme tourne vers 'rien'.
Une fois le programme exécuté vous obtiendrez plusieurs fichiers. Celui
contenant votre modèle avec les nouvelles probabilités estimées se nomme
sigma_seq.model (si votre fichier séquence s'appelle
sigma_seq.txt, sinon l'extension .model remplacera l'extension .txt).
Vous le trouverez dans répertoire seqdna.
Etape de prédiction des positions des régions -35 et -10 en utilisant votre modèle :
Pour réaliser ces prédictions, l'algorithme de Viterbi implémenté dans
SHOW va être utilisé. Nous allons ici tester notre modèle sur les mêmes
séquences que celles utilisées pour l'apprentissage des paramètres. En
général, d'autres séquences sont utilisées lors de l'étape test. Le
fichier devra avoir le même format que sigma_seq.txt.
Il faut se mettre dans la directory contenant les fichiers séquences et votre fichier sigma_seq.txt.
Il faudra lancer la commande suivante :
../bin/show_viterbi -model ../sigma_seq.model -vit ../sigma_vit -seq sigma_seq.txt
le fichier sigma_vit vous a été donné dans l'archive pour l'installation de
SHOW. Il contient les paramètres pour la gestion de la mémoire.
Le programme va créer un fichier .vit par séquence. L'entête du
fichier récapitule les états de votre modèle HMM et attribue un numéro à
chaque état, le premier recevant le chifrre 0, le second le chiffre 1
etc... Les lignes suivantes donnent la valeur numérique associée à
l'état dans lequel la position de la séquence a été prédite.
Exemple d'un extrait d'un fichier .vit :
# viterbi reconstruction
# 0 : (bound) 1 : (background_1) 2
: (sigma-35+_1) 3 : (sigma-35+_2) 4
: (sigma-35+_3) 5 : (sigma-35+_4)....
1
1
1
1
2
3
4
5
6
7
8
9
...
La sortie n'est donc pas directement exploitable pour calculer les
performances de prédiction du modèle, c'est à dire le pourcentage de
fois où le modèle a correctement identifié les boîtes -35 et -10 des
promoteurs sigma A (première position prédite de chacune de ces boîtes
correspond bien aux positions connues dans les séquences analysées).
Pour pouvoir estimer le pouvoir prédictif du modèle, il va falloir parser ces ichiers.
Le programme vous permettant de parser les résultats du viterbi vous est fourni ici. Le sauvegarder dans la directory show_etudiant.
Si le fichier n'apparaît pas comme exécutable, changer son mode. Le
programme prend comme argument en entrée le nom du répertoire contenant
les fichiers séquences, donc ici seqdna.
Un peu de souplesse a été acceptée pour considérer qu'une région avait
été bien prédite. On accepte un écart < 3 entre position réelle et
prédite. Le programme vous affichera les erreurs à l'écran, et donnera
le pourcentage à la fin. Rediriger la sortie à l'écran dans un fichier
pour garder les résultats.
Ce programme peut être largement amélioré et généralisé.
