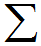 i=1 à 4 Pi log2Pi
i=1 à 4 Pi log2Pi
Exemple, nous avons observé:
| -35 | -10 | |||||||||||
| A | C | G | T | A | C | G | T | |||||
| pos1 | 6 | 3 | 4 | 87 | 3 | 3 | 1 | 93 | ||||
| pos2 | 8 | 4 | 5 | 83 | 95 | 0 | 1 | 4 | ||||
| pos3 | 4 | 7 | 76 | 13 | 23 | 12 | 5 | 59 | ||||
| pos4 | 64 | 12 | 6 | 18 | 77 | 4 | 6 | 13 | ||||
| pos5 | 24 | 51 | 8 | 17 | 74 | 14 | 6 | 7 | ||||
| pos6 | 53 | 9 | 11 | 27 | 4 | 2 | 1 | 93 | ||||
Ces tables s'expriment de la façon suivante avec Patscan:
{(6,3,4,87),(8,4,5,83),(4,7,76,13),(64,12,6,18), (24,51,8,17),(53,9,11,27)}
> 350
16...35
{(3,3,1,93),(95,0,1,4),(23,12,5,59),(77,4,6,13), (74,14,6,7),(4,2,1,93)}
> 400
Les seuils de 350 et 400 ne sont pas optimisés.
i=1 à 4 Pi log2Pisur un ensemble de 4 symboles (les 4 bases). Pi est la fréquence de la base i dans l'ensemble du génome. Si nous avons observé la fréquence Pij de la base i à la position j d'un signal alors l'incertitude du signal Hsj à la position j devient:
i=1 à M Pij log2PijLes probabilités Pij sont estimées par les fréquences des bases observées à chaque position du signal sur un ensemble d'exemples. L'insertitude sera maximum aux positions ne presentant aucune contrainte et sera diminuée aux positions correspondant à un signal. Une façon de mesurer ce biais est de comparer l'insertitude sur l'ensemble du génome (Hg) à l'incertitude au niveau du signal (Hsj):
Rsj = Hg - Hsj
Dans le cas de l'étude d'un ensemble de séquences particulières, il est nécessaire d'introduire une correction qui sera fonction de la taille de l'échantillon et de la fréquence des bases dans cette échantillon. Cette correction consiste à remplacer Hg par l'espérance mathématique de l'entropie Hnb.
Rsj = E(Hnb) -
i=1 à M Pij log2Pij
Le contenu global en information d'un signal est obtenu en sommant sur la longueur du signal (K):
Rs =
j=1 à K Rsj
Pour plus d'information aller sur la page de WebLogo.
j=1 à 4 Fjlog2 [Fj/ Pj]ou Fj est la fréquence observée de la base j à la position i dans toutes les séquences et Pj est la fréquence de cette base dans l'ensemble du génome. Le rapport Fj/ Pj est une mesure de l'écart entre fréquences observée et attendue. Le logarithme en base 2 a été choisi de manière à exprimer l'information en bits. Un raisonement probabiliste basé sur le maximum de vraissemblance conduit à la même formulation (Stormo, 1990).
Matrice déduite:
| -35 | -10 | |||||||||||
| A | C | G | T | A | C | G | T | |||||
| pos1 | -2.22 | -2.874 | -2.46 | 1.64 | -3.22 | -2.874 | -4.46 | 1.73 | ||||
| pos2 | -1.81 | -2.459 | -2.14 | 1.57 | 1.75 | -4.474 | -4.47 | -2.82 | ||||
| pos3 | -2.81 | -1.652 | 1.79 | -1.11 | -0.27 | -0.860 | -2.12 | 1.09 | ||||
| pos4 | 1.19 | -0.874 | -1.87 | -0.64 | 1.40 | -0.709 | -1.93 | -2.06 | ||||
| pos5 | -0.22 | 1.213 | -1.46 | -0.72 | 1.39 | -0.666 | -1.89 | -2.01 | ||||
| pos6 | 0.92 | -1.290 | -1.00 | -0.05 | -2.81 | -3.459 | -4.46 | 1.73 | ||||
Ces matrices deux matrices peuvent s'exprimer de la façon suivante avec Patscan:
{(-22,-29,-25,16),(-18,-25,-21,16),(-28,-17,18,-11),(12,-9,-19,-6),
(-2,12,-15,-7),(9,-13,-10,-1)}
{(-32,-29,-45,17),(17,-45,-45,-28),(-3,-9,-21,11),(14,-7,-19,-21), (14,-7,-19,-20),(-28,-35,-45,17)}
Faire la recherche en utilisant comme seuil 50 pour chacun des motifs.
D =
i=1 à M ln [Pmaxi + 1/N / Pji + 1/N]
ou Pmaxi est la fréquence de la base la plus fréquente
à la position i du signal et Pji est la fréquence
de la base (j) observée à cette position dans le signal testé.
Le facteur 1/N est ajouté à la formule pour éviter une
division par 0 si Pji = 0. Plus la valeur de D est élevée
plus la séquence étudiée est éloingnée
de la séquence consensus. Cet indice s'est avéré très
fortement corrélé (0.88) à l'efficacité des séquences
promotrices mesurées in vitro.
Il a été observé que cet indice donnait le même
poids à toutes les positions du signal, il a donc été
proposé de pondérer chaque position par son contenu en information
(O'Neill, 1989):
D' =
i=1 à M {(E(Hnb) + Pi log2Pi)
(ln [Pmaxi + 1/N / Pji + 1/N])}
Avec cet indice et sur le même ensemble de promoteurs le coeeficient de corrélation monte à 0.97.