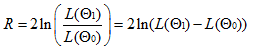Jeux de données
Le programme BlastP a
été utilisé contre les
protéines déduites des gènes identifiés sur les génomes complets avec comme
séquences sondes les protéines ComE et ComD
de S. pneumoniae.
Nous avons utilisé une valeur seuil de e-value de 1e-05.
Nous avons réalisé des alignements et des arbres
afin d'éliminer les séquences partielles ou
très divergentes des séquences ComE ou ComD. Les
séquences retenues ont été extraites
et sont collectées dans deux fichiers au format FASTA
: ComD
et ComE.
Nous n'avons pas retenu de séquences dans les génomes de S. agalactiae et S. suis. Les informations associées à ces
séquences sont compilées dans le tableau 1 (format csv ou en format Excel). Ce
tableau est trié par groupes taxonomiques, par nom
d'espèce et par position des gènes sur le
chromosome. En l'absence de données
expérimentales, nous prédisons que les
partenaires d'un même système (HK et RR) sont
codés par des gènes voisins sur le chromosome.
Afin de faciliter l'étude de la coévolution des
partenaires des systèmes, nous avons exclu des
fichiers FASTA les systèmes incomplets.
En plus du système, Com, les régulons
Blp et Fas ont été étudiés
expérimentalement.
- Blp est impliqué dans la
production de bactériocines. Un petit peptide inducteur (BlP) est codé
par le gène blpC.
Il est probablement maturé et exporté par le
transporteur ABC BlpAB. En réponse au BlP, l'histidine
kinase BlpH s'auto-phosphoryle et transmet son phosphate au régulateur de réponse BlpR.
- Fas (fibronectin/fibrinogen binding/haemolytic activity/streptokinase regulator) a été identifié chez Streptococcus pyogenes. Le système se compose d'un régulateur de réponse (FasA), de deux histidines kinases
(FasB et FasC) et probablement d'un petit ARN non transcrit. Ce
système ne serait pas impliqué dans la régulation
par quorum sensing mais dans la régulation de l'expression de
facteurs de virulences chez les Streptococcus pathogènes du groupe A (GAS).
Les séquences de Staphylococcus epidermidis
et Staphylococcus aureus seront utilisées pour enraciner les arbres.
Analysez le tableau 1. Quelles observations pouvez-vous faire?
Alignement multiples des séquences homologues a ComE de S. pneumoniae
Réaliser
l'alignement multiple des séquences homologues
à ComE avec le programme muscle
au travers du logiciel seaview.
Dans
le menu Align sélectionner Align all
(vérifier le programme sélectionné
dans Alignment options), une nouvelle
fenêtre s'ouvre avec la progression de l'alignement, cliquer sur OK
pour accepter l'alignement dans l'éditeur.
Vérifier
la qualité de l'alignement et effectuer des
corrections manuellement
si nécessaire. Puis sauvegarder l'alignement au format
Fasta.
Attention au nom de fichier, pas d'espace, d'accentuation etc. Conseil
: ComE_muscle.fst;
Construction des arbres en utilisant la
méthode de distance BioNJ
Distance Poisson
Dans
le menu Trees, sélectionner Distance Methods, choisir
la méthode BioNJ
et la distance Poisson.
Vérifier que l'option ignore
all gap sites est cochée. Vous pouvez demander une analyse Bootstrap avec 100 itérations.
L'arbre calculé s'affiche dans un
éditeur d'arbre. Vous pouvez choisir entre plusieurs
représentations et afficher ou non les longueurs de branches
et les valeurs de bootstrap.
- Re-root permet d'enraciner l'arbre à l'aide d'un sous-arbre ou d'une séquence (selectionner le noeud correspondant).
- Swap permet de faire tourner autour d'un noeud, les deux sous-arbres définis par ce noeud.
Pour les autres options se référer au menu Help.
Sauvegarder votre arbre avec le menu File, Save rooted tree. Vous pouvez également utiliser l'option Save to Trees menu pour conserver l'arbre avec le fichier alignement (il apparaîtra dans le menu Trees).
Format Newick
Ouvrir le fichier arbre avec un éditeur de
texte. Vous
découvrirez comment les arbres sont codés en
informatique, le format Newick
qui est utlisé par la plupart des
programmes de phylogénie et interprété
pour vous
fournir une visualisation graphique d'arbre. Les groupes de
séquences sont séparés par des
parenthèses,
les différents groupes par des virgules, le point virgule
indique la fin de l'arbre. Les longueurs de branches sont
indiquées par des valeurs
précédées de deux
points et les valeurs de bootstrap sont indiquées
juste
après la parenthèse fermante
délimitant les
groupes.
Distance Kimura
Calculer l'arbre avec la distance Kimura
Editer les arbres (Swap) de façon à ce que l'ordre des feuilles sur les topologies soient les plus proches possibles.
Comparez les deux topologies, en particulier les longueurs des branches, les valeurs de bootstraps et les incongruences?
Comparaison des bipartitions des arbres obtenues avec les distances de Poisson et Kimura
Nous allons rechercher les bipartitions communes à un ensemble
d'arbre. Une bipartition est obtenue par la coupure d'une branche qui
sépare les feuilles en deux sous-ensembles.
Lancer le logiciel R et charger la librairie ape.
library(ape);
Placez-vous dans le répertoire
contenant les fichiers que vous avez sauvegardés (avec la
commande setwd() par exemple). Pour connaître le chemin de votre
dossier. Clic droit sur le dossier. Ouvrir Propriété.
Vous trouverez l'emplacement. Copier/coller dans R. Attention modifier
les \ en /.
exemple : setwd("C:/Users/fichant/Documents/Phylogenie/Phylogenomic/cours-TD/TD_phylo/2012")
La commande read.tree() permet de lire un arbre dans le format Newick et plot.phylo() permet de déssiner cet arbre.
tP <- read.tree(file = "ComE_BioNJ_Poisson_edited.ph");
plot.phylo(tP, show.node.label = TRUE, edge.color = "blue", edge.width = 1, no.margin = FALSE, cex=0.8);
title('BioNJ Poisson');
axisPhylo(side = 1);
Lire l'arbre obtenu avec la distance de Kimura:
tK <- read.tree(file = "ComE_BioNJ_Kimura_edited.ph");
plot.phylo(tK, show.node.label = TRUE, edge.color = "blue", edge.width = 1, no.margin = FALSE, cex=0.8);
title('BioNJ Kimura');
axisPhylo(side = 1);
et calculer l'arbre consensus strict (les partitions communes à tous les arbres),
tC = consensus(tP, tK, p=1);
plot.phylo(tC, cex=0.8, main='Consensus strict between Poisson and Kimura');
Congruence des arbres obtenues avec les distances de Poisson et Kimura
Nous allons utiliser une méthode
graphique qui permet de dessiner les deux arbres en vis-à-vis et
de relier leur feuilles.
ntips = length(tP$tip.label); # nombre de feuilles dans l'arbre
Creer un tableau associant les feuilles à relier entre les deux arbres (ici elles sont toutes identiques)
association<-matrix(ncol=2, nrow=ntips);
association[,1]<-association[,2]<-tP$tip.label;
cophyloplot(tP, tK, assoc=association, show.tip.label=FALSE, col = "red", use.edge.length=TRUE);
Pour améliorer le rendu, nous allons utiliser une version modifiée des fonctions mycophyloplot.R et myplotCophylo2.R. Sauvegardez ces fonctions dans votre répertoire courant et les lire dans R avec les commandes source("mycophyloplot.R") et source("myplotCophylo2.R"). Puis utilisez la commande:
mycophyloplot(tP, tK, assoc=association, show.tip.label=FALSE, col = "red", use.edge.length=TRUE);
layout(matrix(1:2, 1, 2));
plot.phylo(tC, cex=0.6);
title ('consensus strict');
mycophyloplot(tP, tK, assoc=association, show.tip.label=FALSE, use.edge.length=TRUE);
title('Poisson versus Kimura');
layout(1);
Relation entre les distances d'arbres Poisson/ Kimura
La méthode NJ permet de
reconstruire un arbre à partir d'une matrice de distances. Afin
de comparer les distances inférées sur les arbres, nous
allons extraire et les comparer pour les deux méthodes de
distances.
mDisP <- cophenetic(tP);
mDisK <- cophenetic(tK);
Les matrices sont obtenues dans l'ordre
des feuilles des arbres, or celui peut varier entre les deux arbres.
Comme nous voulons calculer une corrélation entre les paires de
distances calculées avec les deux méthodes, nous devons
tout d'abord trier les matrices de distances mDisP et mDisK sur
les lignes et les colonnes par rapport à l'ordre
alphabétique du nom des séquences. Par cette
procédure, seules les valeurs des matrices ont été
réordonnées mais pas les noms des lignes et des colonnes.
mDisP[rank(colnames(mDisP)),] <- mDisP[c(1:nrow(mDisP)),]; # tri sur les noms des colonnes de mDisP;
mDisP[,rank(rownames(mDisP))] <- mDisP[,c(1:ncol(mDisP))]; # tri sur les noms des lignes de mDisP;
mDisK[rank(colnames(mDisK)),] <- mDisK[c(1:nrow(mDisK)),]; # tri sur les noms des colonnes de mDisK;
mDisK[,rank(rownames(mDisK))] <- mDisK[,c(1:ncol(mDisK))]; # tri sur les noms des lignes de mDisK;
Comme ces matrices sont symétriques
par rapport à la diagonale et que les valeurs de la diagonale
sont nulles, nous allons utiliser la fonction as.dist()
pour extraire la moitié inferieure gauche de la matrice de
distance et éviter ainsi de prendre en compte deux fois les
mêmes paires de valeurs ainsi que les valeurs contenues sur la
diagonale.
plot(as.dist(mDisP),as.dist(mDisK), pch=20, xlab='Poisson', ylab='Kimura');
abline(0,1, col='red');
Construction des arbres en utilisant une méthode du maximum de vraisemblance
Utiliser la méthode
PhyML
implémentée dans seaview avec les
paramètres par défaut.
Comparer le nouvel arbre avec ceux obtenus précédemment avec la méthode
de distance. Voyez-vous des différences? Si oui, cela est-il
étonnant ?
Relation entre les distances d'arbres NJ obtenu avec le modèle Kimura et PhyML obtenu avec le modèle LG
library(ape);
tK <- read.tree(file = "ComE_BioNJ_Kimura_edited.ph");
tLG <- read.tree(file = "ComE_PhyML_LG_edited.ph");
mDisK <- cophenetic(tK);
mDisK[rank(colnames(mDisK)),] <- mDisK[c(1:nrow(mDisK)),];
mDisK[,rank(rownames(mDisK))] <- mDisK[,c(1:ncol(mDisK))];
mDisLG <- cophenetic(tLG);
mDisLG[rank(colnames(mDisLG)),] <- mDisLG[c(1:nrow(mDisLG)),];
mDisLG[,rank(rownames(mDisLG))] <- mDisLG[,c(1:ncol(mDisLG))];
plot(as.dist(mDisK),as.dist( mDisLG), pch=20, xlab='Kim', ylab='LG', main='cophenetic distances');
abline(0,1, col='red');
Présentez les deux arbres en vis-à-vis
layout(matrix(1:2, 1, 2));
plot.phylo(tK, cex=0.6);
title('NJ Kim');
plot.phylo(tLG, cex=0.6, direction='leftwards');
title('PhyML LG');
layout(1);
Comparaison des bipartitions des trois arbres
tP <- read.tree(file = "ComE_BioNJ_Poisson_edited.ph");
tK <- read.tree(file = "ComE_BioNJ_Kimura_edited.ph");
tLG <- read.tree(file = "ComE_PhyML_LG_edited.ph");
tC <- consensus(tP, tK, tLG, p=1);
plot.phylo(tC, cex=0.8);
title('Consensus strict, Poisson, Kimura, PAM, and LG');
Coloriage de l'arbre en fonction de la classe des protéines
Lecture du tableau en format csv
tableau <- read.table("Gene_description_CleanUp-1.csv", header=TRUE, sep=';')
Code couleur pour chaque classe:
codes <- matrix(c('COME', 'BLPR', 'FASA', 'blue', 'red', 'green'), 3,2)
Association de la classe de la protéine (colonne Classification) à une couleur en gardant l’ordre des feuilles dans l’arbre tC.
col <- codes[match(tableau[match(tC$tip, tableau$Gene_Name), 11], codes[,1]), 2]
plot.phylo(tC, cex=0.8, tip.color=col);
legend("bottomleft", legend=codes[,1], col=codes[,2], pch=20)
Concatener les arbres dans un seul fichier:
new_file <- "Concatenated_edited.ph";
write.tree(tP, file = new_file, append = FALSE);
write.tree(tK, file = new_file, append = TRUE);
write.tree(tLG, file = new_file, append = TRUE);
Edition et annotation des arbres
Nous allons utiliser le programme treedyn
pour comparer nos arbres.
- Lancer le programme treedyn.
- Dans le menu File
choisir Open tree(s) pour selectionner l'arbre à charger
(le fichier avec les arbres concaténés).
- Puis choisir Load
Annotations et charger le fichier Gene_description.tlf
que vous avez enregistré sur votre ordinateur.
- Dans Load
Script charger le fichier Gene_description.tds
que vous avez enregistré sur votre ordinateur.
- Cliquer sur les arbres (clic droit) de droite et
selectionner Conformation
-> Mirror.
- Replacer les arbres en vis-à-vis.
- Vous pouvez ajouter les valeurs de bootstrap avec l'option Display Newick Annotations accessible dans le menu otenu par un clic droit dans une zone contenant un arbre. Le choix X: correspond aux valeurs de bootstrap et et le choix :X aux longueurs de branches.
- Vous donner un nom a chaque arbre avec l'option Name Tree du menu arbre et ajouter ces noms avec l'option Add Tree(s) Names du menu général.
Comparer et discuter les quatre arbres.
Effet des paramètres de PhyML sur la
reconstruction des arbres
Vitesse
différentes d'évolution des sites : nombre de
classes de sites
Par
défaut le nombre de classes de site est fixé
à 4. Essayer 8 classes. Commentaires.
tLG8 <- read.tree(file = "ComE_PhyML_LG8_edited.ph");
tC = consensus(tLG, tLG8, p=1);
plot.phylo(tC);
mDisLG8 <- cophenetic(tLG8);
mDisLG8[rank(colnames(mDisLG8)),] <- mDisLG8[c(1:nrow(mDisLG8)),];
mDisLG8[,rank(rownames(mDisLG8))] <- mDisLG8[,c(1:ncol(mDisLG8))];
plot(as.dist(mDisLG),as.dist( mDisLG8), pch=20, xlab='LG 4', ylab='LG 8');
title('cophenetic distances');
abline(0,1, col='red');
ntips = length(tLG$tip.label);
association<-matrix(ncol=2, nrow=ntips);
association[,1]<-association[,2]<-tLG$tip.label;
layout(matrix(1:4, 2, 2));
col <- codes[match(tableau[match(tC$tip, tableau$Gene_Name), 11], codes[,1]), 2]
plot.phylo(tC, cex=0.5, tip.color=col);
title('consensus strict');
col <- codes[match(tableau[match(tLG$tip, tableau$Gene_Name), 11], codes[,1]), 2]
plot.phylo(tLG, cex=0.5, tip.color=col);
title('LG 4');
mycophyloplot(tLG, tLG8, assoc=association, use.edge.length=TRUE);
title('LG 4 versus LG 8');
col <- codes[match(tableau[match(tLG8$tip, tableau$Gene_Name), 11], codes[,1]), 2]
plot.phylo(tLG8, cex=0.5, direction='leftwards', tip.color=col);
title('LG 8');
layout(1);
Recherche du modèle évolutif
le plus adapté à nos données
Nous allons utiliser le programme prottest3
qui permet de selectionner le meilleur modéle d'évolution pour
un alignement de séquences d'acides aminés. Le
programme est disponible comme ressource WEB (prottest2_server) ou comme application que vous pouvez lancer sur votre PC. Nous allons utiliser la version locale que l'on peut obtenir en allant sur le site drive.google.com pour télécharger l'archive (dernière version, cliquez sur la flèche vers le bas dans le bandeau).
Décompresser l'archive à l'aide du programme 7-Zip (clic
droit sur le nom du fichier, extraire ici: la première
opération vous décompresse le fichier; on obtient le
fichier avec l'extension .tar. recommencer la même
opération sur ce nouveau fichier. Cela vous extrait vos fichier
de l'archive et vous construit le répertoire prottest3). Aller
dans le répertoire prottest3 et lancer la commande runXProtTestHPC.bat.
- menu File selectionner l'alignement à analyser (format FASTA).
- menu Analysis, choisir Compute likelihood scores,
sélectionner les matrices JTT (Jones et al., 1992), LG (Le and Gascuel, 2008), WAG (Whelan and Goldman, 2001) et Blosum62 (Henikoff and Henikoff, 1992) et l'estimation des
vitesses de variations (+G) avec 4 catégories et des fréquences des acides aminés (+F). Puis lancer la
recherche...
- menu Selection, choisir Results.
Vous avez un tableau avec les différents modèles
classés par ordre décroissant par rapport à leur
adéquation avec l'alignement multiple utilisé.
- Utiliser Export to main console pour copier les résultats dans la console et les enregistrer dans un fichier.
Dans le tableau Results vous avez plusieurs calculs
réalisés suivant différents critères,
notamment l'AIC qui signifie Akaike
Information Criterion (AIC).
Lorsque l'on estime un modèle statistique, il est possible d'augmenter la vraisemblance du modèle en ajoutant un paramètre. Le critère d'information d'Akaike permet de pénaliser les modèles en fonction du nombre de paramètres afin de satisfaire le critère de parcimonie. On choisit alors le modèle avec le critère d'information d'Akaike le plus faible.
Dans notre contexte, c'est
un
estimateur qui correspond à la minimisation de la distance
attendue entre un modèle vrai et son estimation. Les
modèles correspondant aux valeurs minimales de l'AIC sont
considérés comme les plus appropriés
pour la
reconstruction. Une même topologie de
référence
doit être utilisée pour tester les
différents
modèles.
Le critère d'information d'Akaike s'écrit comme la différence entre 2 fois le nombre de paramètres (k) deux fois la log-vraisemblance du modèle estimé.
k = nombre de
paramètres libres du modèle
Alignement
multiples des séquences homologues a ComD de S. pneumoniae
Réaliser
l'alignements multiples des séquences homologues
à ComD avec le programme muscle
au travers du logiciel seaview comme vous l'avez fait pour ComE.
Vérifier
la qualité de l'alignements et effectuer des
corrections manuellement
si nécessaire. Puis sauvegarder l'alignement.
Recherche du modèle évolutif
le plus adapté à nos données
Utiliser prottest3 pour trouver le modèle le mieux adapté à ces séquences.
Construction de l'arbre en utilisant une
méthode du maximum de vraisemblance
Utiliser la méthode
PhyML
implémentée dans seaview avec les
paramètres par défaut.
Comparaison des arbres avec R
tComE <- read.tree(file = "ComE_PhyML_LG_edited_ComD.ph");
tComD <- read.tree(file = "ComD_PhyML_LG_edited_ComE.ph");
codes2 <- matrix(c('COMD', 'BLPH', 'FASB', 'FASC', 'blue', 'red', 'green', 'lightgreen'), 4,2)
col2 <- codes2[match(tableau[match(tComD$tip, tableau$Gene_Name), 11], codes2[,1]), 2]
layout(matrix(1:2, 1,2));
plot.phylo(tComE, cex=0.5, tip.color=col);
legend("topleft", legend=codes[,1], col=codes[,2], pch=20, cex=0.8)
title('ComE');
plot.phylo(tComD, cex=0.5, direction='leftwards', tip.color=col2);
legend("topright", legend=codes2[,1], col=codes2[,2], pch=20, cex=0.8)
title('ComD');
layout(1);
Construction d'un arbre espèces en utilisant un ensemble de protéines
Une des approches utilisées pour reconstruire
l'arbre phylogénétique d'un ensemble d'espèces consiste à concaténer les
alignements obtenus sur un ensemble de gènes ou de protéines conservés dans les
génomes de ces espèces (méthode supermatrice). Nous allons illustrer cette
approche avec les protéines des petites et grandes sous-unités des ribosomes.
Nous avons utilisé les profils COGs de la base de données CD pour annoter les
génomes complets de streptocoques à l'aide du programme RPS-BLAST. Nous avons
retenu un échantillon de 43 familles de protéines (tableau 2) qui étaient annotées correctement dans la grande
majorité des 17 génomes complets de streptocoques. Les séquences du
génome de Lactococcus lactis seront utilisées pour enraciner l'arbre.
Pour chaque famille, les séquences des protéines ont été extraites (Ribo_proteins).
Illustration
A titre d'illustration, nous allons utiliser seaview
pour aligner avec muscle les 5 premiers fichiers et nous allons
calculer les arbres phylogénétiques sur ces fichiers.
Pour pouvoir réaliser facilement l'étape de concaténation, pensez
à ouvrir vos fichiers à partir de la première
fenêtre seaview. Dans tous les cas sauvegarder vos alignements.
Que pensez-vous des alignements obtenus?
Que pensez-vous des arbres obtenus?
Recherchons les bipartitions communes à tous ces arbres
t1 <- read.tree(file = "Tree1.ph");
t2 <- read.tree(file = "Tree2.ph");
t3 <- read.tree(file = "Tree3.ph");
t4 <- read.tree(file = "Tree4.ph");
t5 <- read.tree(file = "Tree5.ph");
tC = consensus(t1, t2, t3, t4,t5, p=1);
Qu'avez vous obtenus?
Supprimer l'arbre qui pose des problèmes et recommencer l'opération.
plot.phylo(tC, cex=0.8, main = 'Consensus strict');
Nous allons maintenant concaténer les 5 premiers alignements.
Pour cela, placez-vous dans la fenêtre seaview contenant le
premier alignement et dans le menu File choisissez Concatenate.
On selectionne successivement les 4 autres alignements. Pensez à cocher la case add gaps pour que les alignements avec un nombre de séquences plus petit soient correctement entrés.
Que pensez-vous de l'alignement obtenu?
Nous allons utiliser le pgrogramme Gblocks
pour éliminer automatiquement les positions avec une faible ou
aucune information phylogénétique. Dans le menu Sites choisir Create set, puis Gblocks. Dans le menu Gblocks cocher Allow gap positions within final blocks.
Lancer le calcul de l'arbre phylogénétique.
Vous pouvez utiliser treedyn pour annoter votre arbre (Species_description.tlf et Species_description.tds).
Alignement des 43 familles de
protéines
Dans le fichier (super matrix) vous trouverez la concaténation des alignements des 43 familles de
protéines.
Calculer un arbre en utilisant la méthode BioNJ et la matrice de distance Kimura (100 bootstraps) et un arbre en utilisant PhyML et le modèle LG avec G = 4 et aLRT (compter ~ 15 min.).
Construction de la phylogénie des
Streptocoques à partir des séquences de l'ARN 16S
Nous
allons réaliser cette nouvelle phylogénie
à l'aide des séquences des ARNr 16S. Nous allons
utiliser
comme
ressource la Ribosomal Database
Project II (RDP).
Plusieurs outils d'analyses vous sont proposés.
Utiliser
le Hierarchy browser (lien Browsers) pour
sélectionner les
séquences d'ARNr 16S des streptococcus.
Sélectionner
seulement les séquences types.
Combien de séquences obtenez_vous?
Cocher toutes les
séquences pour les sélectionner (cliquer dans le
+).
Ensuite faire download
pour
qu'elles soient mises dans votre seqCart.
On peut choisir en plus de sauvegarder ces séquences sur son
ordinateur. Pour cela, choisir le format Fasta et garder l'option
Remove common gaps sinon nous obtenons un alignement très
long
plein de gaps. Ouvrir le fichier avec Seaview (format Fasta). Vous
allez voir que vos séquences sont identifiées
suivant
leur accession number dans la RDP et que vous avez donc perdu le nom de
l'espèce. Il va donc falloir les renommer. Comme cela est
fastidieux, vous trouverez ici
le fichier à utiliser.
Vous pouvez soit
construire
l'arbre directement dans Seaview avec la distance HKY (la plus proche
de Tajima & Nei), ou ouvrir ce dernier dans Seaview
et sauvegarder en format mase et
réaliser l'arbre phylogénétique avec
phylo_win en
utilisant la distance de Tajima & Nei et la Neigbor Joining
method.
Les calculs étant assez rapides vous pouvez tenter 50
bootstrap.
Votre topologie est-elle bien résolue?
Nous allons suppimer les séquences d'ARNr 16S qui sont
redondantes pour certains organismes. Refaire l'arbre. Etes-vous
satisfait de votre topologie.
- Recherche du modèle évolutif
le plus adapté à nos données.
Etant
donné le grand
nombre de modèles évolutifs disponibles pour le
traitement des séquences d'acides nucléiques, des
méthodes ont été
développées pour
permettre de choisir le modèle le plus adapté aux
données. Elles utilisent PhyML comme méthode de
reconstruction d'arbre. Deux tests seront utilisés :
- Le
rapport de vraisemblance ou LRT (Likelihood Ratio Test)
Deux
modèles sont
comparés (modèle correponsdant à
l'hypothèse nulle et le modèle alternatifs) et le
rapport
des vraisemblances est calculé.
Le rapport de vraisemblance est d'autant plus grand que la
vraisemblance du modèle alternatif est grande. Le
modèle
nul sera rejeté si R est supérieur au seuil
fixé
par l'utilisateur. Pour que le LRT est un sens il faut que les
modèles soient imbriqués car parcours d'un arbre
de
décision et testé sur une même
topologie de
référence.
Exemple d'un arbre de décision (extrait du manuel de
JModeltest).
- L'Akaike
Information Criterion (AIC)
C'est une
alernative au LRT
qui présente l'avantage de pouvoir être
appliquée
à des hypothèses non imbriquées. C'est
un
estimateur qui correspond à la minimisation de la distance
attendue entre un modèle vrai et son estimation. Les
modèles correspondant aux valeurs minimales de l'AIC sont
considérés comme les plus appropriés
pour la
reconstruction. Une même topologie de
référence
doit être utilisée pour tester les
différents
modèles.
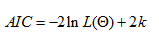 k = nombre de
paramètres libres du modèle
k = nombre de
paramètres libres du modèle
Nous
allons donc rechercher
le modèle le plus approprié correspondant
à nos
données sur ComD pour réaliser notre arbre. Aller
sur le
site de
Phylemon2.
Créer un compte ou se connecter en tant
qu'utilisateur anonyme. Rechercher l'application
JMoldelTest.
Copier/coller votre alignement. Pour un nombre de type de substitution
égale à 3, utiliser les deux méthodes
AIC et LRT.
Pour cette dernière on effectuera l'option " Do dynamical
likelihood ratio tests".
Quel modèle est le plus adéquate?
A quoi correspond ce modèle ? (voir le manuel de JModelTest
dans le Help)
Est-il implémenté dans PhyML?
Quel est le second modèle le plus adapté?
Noter l'impact de la correction par la gamma distribution sur la
vraisemblance du modèle.
Reconstruire l'arbre en choisissant le modèle
proposé
dans PhyML et en choisissant Best of NNI & SPR pour la
recherche de
l'arbre.
Comparer avec le précédent. La topologie est-elle
bien résolue?
Nous allons donc essayer d'améliorer celle-ci en recherchant
de
façon exhaustive le modèle le plus
approprié. Pour
cela sur Phylemon2 rechercher l'option
Phyml
Best AIC Tree (v. 1.02b). Sauveragder l'arbre obtenu dans un
fichier et importer le ensuite dans seaview.
Persiste-t-il encore des problèmes de résolution
des branches?
 Approche génomique : étude de l'évolution de la
cascade de régulation de la compétence chez les
Streptocoques
Approche génomique : étude de l'évolution de la
cascade de régulation de la compétence chez les
Streptocoques