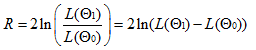Exercice 1 : Construction des arbres sur les protéines homologues à ComD et ComE par une méthode de distance.
Vous trouverez ici les sélections proposées des protéines homologues à ComD et à ComE.
Pour chacun des deux jeux de données réaliser les
alignements multiples avec le programme muscle au travers du logiciel
seaview puis sauvegarder vos alignement dans deux formats
différents Fasta et Mase (ce format est utilisé par le
programme phylo_win). Attention au nom de fichier, pas d'espace,
d'accentuation etc. Phylo_win est très susceptible !
Conseil : ComD_muscle.fst et ComD_muscle.mase.
Même si le programme phylo_win n'est plus maintenu et
remplacé par seaview nous allons l'utiliser pour pouvoir
réaliser un arbre en utilisant le modèle évolutif
PAM pour calculer les distances entre nos séquences.
Installation du program phylo_win dans votre répertoire de travail. Aller sur le serveur lyonnais
PBIL et télécharger la version windows de phymo_win. L'installer.
- Construire un arbre de parenté pour chaque jeu de
données en utilisant la méthode de distance NJ avec une
distance PAM et 10 bootstrap (pas plus car temps de calcul assez long).
Sauvegarder votre arbre dans un fichier (option Tree File dans le menu
Output). Donner un nom de fichier avec une extension .ph (ex :
Tree_ComD_PAM.ph).
- Ouvrir le fichier avec un éditeur de texte. Vous
découvrirez comment les arbres sont codés en
informatique, le format Newick qui est utlisé par la plupart des
programmes de phylogénie et interprété pour vous
fournir une visualisation graphique d'arbre. Les groupes de
séquences sont séparés par des parenthèses,
les différents groupes par des virgules, le point virgule
indique la fin de l'arbre. Les longueurs de branches sont
indiquées par des valeurs précédées de deux
points et les valeurs de bootstrap sont indiquées juste
après la parenthèse fermante délimitant les
groupes.
- Importer chacun des arbres dans seaview et grace à
l'éditeur d'arbre enraciner les correctements et travailler la
mise en forme des topologies de manière à pouvoir
comparer facilement les deux arbres (bouton swap mettre les groupes de
séquences dans le même ordre si possible).
- Comparer et discuter les deux arbres. Conclusion biologique?
Exercice 2 : Construction des arbres en utilisant une méthode du maximum de vraisemblance.
Refaire vos deux arbres en utilisant la PhyML
implémentée dans seaview. On utilisera les
paramètres par défaut. Comparer à ceux obtenus
avec la méthode
de distance. Voyez-vous des différences? Si oui, cela est-il
étonnant?
Exercice 3 : Analyse de l'effet des paramètres sur la reconstruction d'arbre de séquences protéiques.
- Effet du modèle évolutif.
Nous travaillerons avec le jeu
de données de ComD. Reconstruire l'arbre avec PhyML mais en
chosissant le modèle JTT à la place du modèle LG.
Quelle(s) différence(s) voyez vous entre les deux arbres
(topologie, longueurs des branches). Lequel des deux arbres est-il le
plus vraisemblable? Quel modèle est donc le plus
approprié à vos données?
- Vitesse différentes d'évolution des sites : nombre de classes de sites
Par défaut le nombre de classes de site est fixé à 4. Essayer 8 classes. Commentaires.
Exercice 4 : Construction de la phylogénie des Streptocoques
Pour réaliser cette phylogénie nous allons utiliser
les séquences des ARNr 16S. Nous allons utiliser comme
ressource la Ribosomal Database
Project II (RDP).
Plusieurs outils d'analyses vous sont proposés. Utiliser
le Hierarchy browser (lien Browsers) pour
sélectionner les
séquences d'ARNr 16S des streptococcus. Sélectionner
seulement les séquences types.
Combien de séquences obtenez_vous?
Cocher toutes les
séquences pour les sélectionner (cliquer dans le +).
Ensuite faire download pour
qu'elles soient mises dans votre seqCart.
On peut choisir en plus de sauvegarder ces séquences sur son
ordinateur. Pour cela, choisir le format Fasta et garder l'option
Remove common gaps sinon nous obtenons un alignement très long
plein de gaps. Ouvrir le fichier avec Seaview (format Fasta). Vous
allez voir que vos séquences sont identifiées suivant
leur accession number dans la RDP et que vous avez donc perdu le nom de
l'espèce. Il va donc falloir les renommer. Comme cela est
fastidieux, vous trouverez ici
le fichier à utiliser.
Vous pouvez soit construire
l'arbre directement dans Seaview avec la distance HKY (la plus proche
de Tajima&Nei), ou ouvrir ce dernier dans Seaview
et sauvegarder en format mase comme à l'exercice 1et
réaliser l'arbre phylogénétique avec phylo_win en
utilisant la distance de Tajima&Nei et la Neigbor Joining method.
Les calculs étant assez rapides vous pouvez tenter 50 bootstrap.
Votre topologie est-elle bien résolue?
Nous allons suppimer les séquences d'ARNr 16S qui sont
redondantes pour certains organismes. Refaire l'arbre. Etes-vous
satisfait de votre topologie.
- Recherche du modèle évolutif le plus adapté à nos données.
Etant donné le grand
nombre de modèles évolutifs disponibles pour le
traitement des séquences d'acides nucléiques, des
méthodes ont été développées pour
permettre de choisir le modèle le plus adapté aux
données. Elles utilisent PhyML comme méthode de
reconstruction d'arbre. Deux tests seront utilisés :
- Le rapport de vraisemblance ou LRT (Likelihood Ratio Test)
Deux modèles sont
comparés (modèle correponsdant à
l'hypothèse nulle et le modèle alternatifs) et le rapport
des vraisemblances est calculé.
Le rapport de vraisemblance est d'autant plus grand que la
vraisemblance du modèle alternatif est grande. Le modèle
nul sera rejeté si R est supérieur au seuil fixé
par l'utilisateur. Pour que le LRT est un sens il faut que les
modèles soient imbriqués car parcours d'un arbre de
décision et testé sur une même topologie de
référence.
Exemple d'un arbre de décision (extrait du manuel de JModeltest).
- L'Akaike Information Criterion (AIC)
C'est une alernative au LRT
qui présente l'avantage de pouvoir être appliquée
à des hypothèses non imbriquées. C'est un
estimateur qui correspond à la minimisation de la distance
attendue entre un modèle vrai et son estimation. Les
modèles correspondant aux valeurs minimales de l'AIC sont
considérés comme les plus appropriés pour la
reconstruction. Une même topologie de référence
doit être utilisée pour tester les différents
modèles.
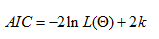 k = nombre de paramètres libres du modèle
k = nombre de paramètres libres du modèle
Nous allons donc rechercher
le modèle le plus approprié correspondant à nos
données sur ComD pour réaliser notre arbre. Aller sur le
site de Phylemon2. Créer un compte ou se connecter en tant qu'utilisateur anonyme. Rechercher l'application JMoldelTest.
Copier/coller votre alignement. Pour un nombre de type de substitution
égale à 3, utiliser les deux méthodes AIC et LRT.
Pour cette dernière on effectuera l'option " Do dynamical
likelihood ratio tests".
Quel modèle est le plus adéquate?
A quoi correspond ce modèle ? (voir le manuel de JModelTest dans le Help)
Est-il implémenté dans PhyML?
Quel est le second modèle le plus adapté?
Noter l'impact de la correction par la gamma distribution sur la vraisemblance du modèle.
Reconstruire l'arbre en choisissant le modèle proposé
dans PhyML et en choisissant Best of NNI & SPR pour la recherche de
l'arbre.
Comparer avec le précédent. La topologie est-elle bien résolue?
Nous allons donc essayer d'améliorer celle-ci en recherchant de
façon exhaustive le modèle le plus approprié. Pour
cela sur Phylemon2 rechercher l'option Phyml Best AIC Tree (v. 1.02b). Sauveragder l'arbre obtenu dans un fichier et importer le ensuite dans seaview.
Persiste-t-il encore des problèmes de résolution des branches?
Certains sites pouvant introduire
du bruit, il peut être nécessaire de ne pas les prendre en
compte lors de l'analyse phylogénétique. Le programme
trimAL permet de "nettoyer" votre alignement. Il est disponible sur
Phylemon2. L'appliquer en choisissant la méthode Automated1
optimisé pour les reconstructions phylogénétique
utilisant le maximum de vraisemblance). Sauvegarder l'alignement et
refaire la reconstruction comme ci-dessus avec l'option Phyml Best AIC Tree (v. 1.02b).
Il nous est difficile d'obtenir une topologie bien résolue car
si nous regardons l'alignement des séquences d'ARNr 16S, la
majorité des sites sont soit très bien conservés,
soit fortement variables.
- Utilisation d'un autre jeu de données.
Utilisation d'un gène de ménage, conservé dans l'ensemble des génomes. Protéines GroeL ( sequences ici).
Faire la reconstruction phylogénétique avec ce nouvel
ensemble de séquences. Optimiser pour les sites invariants. Pour
les étapes de rebranchements lors de la recherche de l'arbre
faire Best of NNI & SPR.
Comparer à l'arbre des ARNr 16S.
On voit qu'il est difficile à partir d'un seul gène
d'obtenir une phylogénie stable. D'où les approches de
type super-arbre et super-matrice qui ont été
développées.
|
 Approche génomique : étude de l'évolution de la
cascade de régulation de la compétence chez les Streptocoques (suite)
Approche génomique : étude de l'évolution de la
cascade de régulation de la compétence chez les Streptocoques (suite)