


M1 MABS BBS Math TD Calcul Matriciel
From silico.biotoul.fr
m (→Matrices de substitution) |
m (→Matrices de substitution) |
||
| Line 41: | Line 41: | ||
| - | = | + | = Application aux matrices de substitution pour les alignements de séquences = |
| + | |||
| + | == Calcul d'une matrice de substitution à partir des alignements == | ||
Nous allons construire une matrice de type Dayhoff (PAM) pour l'alignement de séquences protéiques, ceci à partir d'alignements de séquences. La méthode en quelques mots consiste à | Nous allons construire une matrice de type Dayhoff (PAM) pour l'alignement de séquences protéiques, ceci à partir d'alignements de séquences. La méthode en quelques mots consiste à | ||
| Line 92: | Line 94: | ||
| - | La matrice de substitution se calcule donc à l'aide de la formule suivante : <math>S_{i,j} = 10 | + | La matrice de substitution se calcule donc à l'aide de la formule suivante : <math>S_{i,j} = 10 \times log_{10}\frac{M_{i,j}}{F_i}</math> |
| - | Nous avons donc besoin de calculer la fréquences de chaque acide aminé dans une séquence (ici, fréquences d'apparition dans nos alignements de départ). | + | Nous avons donc besoin de calculer la fréquences de chaque acide aminé dans une séquence (ici, fréquences d'apparition dans nos alignements de départ). Utilisez donc la matrice <tt>CM</tt> de départ qui contient le nombre de substitutions observées pour calculer le vecteur <tt>Freq</tt> contenant la fréquence de chacun de a.a. |
| + | |||
| + | Vous devriez obtenir le vecteur suivant : | ||
| + | <source lang="rsplus"> | ||
| + | round(Freq,2) | ||
| + | A R N D C Q E G H I L K M F P S T W Y V | ||
| + | 0.08 0.05 0.04 0.06 0.01 0.03 0.07 0.08 0.02 0.07 0.09 0.07 0.02 0.04 0.04 0.06 0.06 0.01 0.03 0.08 | ||
| + | </source> | ||
| + | |||
| + | |||
| + | Il n'y a plus qu'à appliquer la formule <math>S_{i,j} = 10 \times log_{10}\frac{M_{i,j}}{F_i}</math>. | ||
| + | |||
| + | Vous devriez obtenir la matrice de substitution suivante : | ||
| + | |||
| + | <source lang="rsplus"> | ||
| + | round(S[1:10,1:10],1) | ||
| + | A R N D C Q E G H I | ||
| + | A 8.1 -5.3 -4.4 -4.5 -0.2 -3.0 -2.3 -2.1 -6.2 -6.0 | ||
| + | R -5.3 10.5 -2.7 -6.0 -4.8 0.5 -2.9 -6.4 -1.6 -8.4 | ||
| + | N -4.4 -2.7 10.9 1.9 -4.5 -1.4 -1.8 -1.8 0.7 -8.3 | ||
| + | D -4.5 -6.0 1.9 10.0 -9.3 -1.7 2.9 -4.5 -2.2 -13.1 | ||
| + | C -0.2 -4.8 -4.5 -9.3 16.6 -8.4 -11.3 -6.7 -6.1 -4.3 | ||
| + | Q -3.0 0.5 -1.4 -1.7 -8.4 10.8 1.8 -6.5 0.1 -8.1 | ||
| + | E -2.3 -2.9 -1.8 2.9 -11.3 1.8 8.8 -5.7 -3.3 -10.5 | ||
| + | G -2.1 -6.4 -1.8 -4.5 -6.7 -6.5 -5.7 9.9 -7.1 -14.3 | ||
| + | H -6.2 -1.6 0.7 -2.2 -6.1 0.1 -3.3 -7.1 14.1 -9.8 | ||
| + | I -6.0 -8.4 -8.3 -13.1 -4.3 -8.1 -10.5 -14.3 -9.8 8.6 | ||
| + | </source> | ||
Revision as of 10:32, 29 September 2011
Contents |
Création d'une matrice
A = matrix( c(11, 12, 13, 21, 22, 23, 31, 32, 33), ncol=3, byrow=TRUE )
A partir d'un fichier File:Gold.metadata.txt :
G=read.table("gold.metadata.txt", sep="\t", header=TRUE) class(G)
G est un data frame ; les deux premières colonnes contiennent l'identifiant et le nom de l'organisme. Pour extraire la matrice de données (colonnes numériques), on fait :
as.matrix( G[ , 3:12] )
Cas de la matrice unité d'ordre n (notée In), exemple avec n = 3 :
I3 = diag(3)
Cas des vecteurs colonne ou ligne :
V = c(1, 0, 5) class(V) # obtention de la matrice vecteur colonne : as.matrix(V) # pour obtenir la matrice vecteur ligne, on fait la transposée du résultat pécédent : t( as.matrix(V) )
Opérations sur les matrices
- addition +, soustraction -, multiplication par un nombre *,
- produit : entre 2 matrices A et I3 : A %*% I3, entre une matrice A et un vecteur V A %*% V
- transposition : t(M)
Application aux matrices de substitution pour les alignements de séquences
Calcul d'une matrice de substitution à partir des alignements
Nous allons construire une matrice de type Dayhoff (PAM) pour l'alignement de séquences protéiques, ceci à partir d'alignements de séquences. La méthode en quelques mots consiste à
- compter le nombre de substitutions observées pour chaque acide aminé (a.a.),
- à transformer ce nombre d'occurrence en fréquence,
- puis à calculer la log odds matrix : la matrice contenant les coûts attribuer à chaque substitution (match et mismatch).
La première étape (déjà réalisée pour vous) consiste à réaliser les alignements et à dénombrer le nombre de passage d'un acide aminé à un autre.
Fichier contenant, le nombre de substitutions observées entre chaque paire d'acides aminés dans un lot de séquences préalablement alignées File:Count matrix.txt.
CM=read.table("count_matrix.txt", sep="\t", header=TRUE, row.names=1)
Après avoir chargé cette matrice dans R, transformez-la en une matrice MM (mutation matrix) qui contient des fréquences (aidez-vous des fonctions apply et sum). Ces fréquences sont interprétées comme des probabilités de passer d'un acide aminé en un autre.
Vous devriez obtenir ceci (10 premières lignes et 10 premières colonne) :
round(MM[1:10,1:10],2) A R N D C Q E G H I A 0.54 0.03 0.03 0.03 0.08 0.04 0.05 0.05 0.02 0.02 R 0.02 0.57 0.03 0.01 0.02 0.06 0.03 0.01 0.04 0.01 N 0.01 0.02 0.48 0.06 0.01 0.03 0.03 0.03 0.05 0.01 D 0.02 0.01 0.09 0.57 0.01 0.04 0.11 0.02 0.03 0.00 C 0.01 0.00 0.00 0.00 0.57 0.00 0.00 0.00 0.00 0.00 Q 0.02 0.04 0.03 0.02 0.01 0.42 0.05 0.01 0.04 0.01 E 0.04 0.04 0.05 0.13 0.01 0.11 0.52 0.02 0.03 0.01 G 0.05 0.02 0.05 0.03 0.02 0.02 0.02 0.77 0.02 0.00 H 0.00 0.01 0.02 0.01 0.01 0.02 0.01 0.00 0.54 0.00 I 0.02 0.01 0.01 0.00 0.03 0.01 0.01 0.00 0.01 0.50
Pour un alignement de l'acide aminé AAi avec l'acide aminé AAj, on utilise la formule suivante :

avec M la matrice MM précédemment calculée (mutation matrix) et F la fréquence de chacun des acides aminés (ici, les fréquences d'apparition dans nos alignements de départ).
On en déduit la vraisemblance d'un alignement entre AAi et AAj : 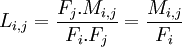
En pratique, on utilise les logarithmes car 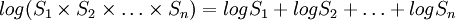 .
.
La matrice de substitution se calcule donc à l'aide de la formule suivante : 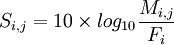
Nous avons donc besoin de calculer la fréquences de chaque acide aminé dans une séquence (ici, fréquences d'apparition dans nos alignements de départ). Utilisez donc la matrice CM de départ qui contient le nombre de substitutions observées pour calculer le vecteur Freq contenant la fréquence de chacun de a.a.
Vous devriez obtenir le vecteur suivant :
round(Freq,2) A R N D C Q E G H I L K M F P S T W Y V 0.08 0.05 0.04 0.06 0.01 0.03 0.07 0.08 0.02 0.07 0.09 0.07 0.02 0.04 0.04 0.06 0.06 0.01 0.03 0.08
Il n'y a plus qu'à appliquer la formule .
Vous devriez obtenir la matrice de substitution suivante :
round(S[1:10,1:10],1) A R N D C Q E G H I A 8.1 -5.3 -4.4 -4.5 -0.2 -3.0 -2.3 -2.1 -6.2 -6.0 R -5.3 10.5 -2.7 -6.0 -4.8 0.5 -2.9 -6.4 -1.6 -8.4 N -4.4 -2.7 10.9 1.9 -4.5 -1.4 -1.8 -1.8 0.7 -8.3 D -4.5 -6.0 1.9 10.0 -9.3 -1.7 2.9 -4.5 -2.2 -13.1 C -0.2 -4.8 -4.5 -9.3 16.6 -8.4 -11.3 -6.7 -6.1 -4.3 Q -3.0 0.5 -1.4 -1.7 -8.4 10.8 1.8 -6.5 0.1 -8.1 E -2.3 -2.9 -1.8 2.9 -11.3 1.8 8.8 -5.7 -3.3 -10.5 G -2.1 -6.4 -1.8 -4.5 -6.7 -6.5 -5.7 9.9 -7.1 -14.3 H -6.2 -1.6 0.7 -2.2 -6.1 0.1 -3.3 -7.1 14.1 -9.8 I -6.0 -8.4 -8.3 -13.1 -4.3 -8.1 -10.5 -14.3 -9.8 8.6