M1 MABS Graphes TP Visualisation et parcours en profondeur
From silico.biotoul.fr
m (→Attributs sur les sommets et arêtes) |
m (→Parcours en profondeur d'un graphe) |
||
| (14 intermediate revisions not shown) | |||
| Line 20: | Line 20: | ||
'''Récupérez''' le fichier [[Media:String_EcolA_coexpression.sif|String_EcolA_coexpression.sif]] contenant une partie du réseau d'interaction basé sur la co-expression chez ''Escherichia coli'' K12 avec un seuil de 0.5 pour la confiance dans une arête et '''ouvrez'''-le dans Cytoscape (menu <tt>File->Import->Network</tt>). | '''Récupérez''' le fichier [[Media:String_EcolA_coexpression.sif|String_EcolA_coexpression.sif]] contenant une partie du réseau d'interaction basé sur la co-expression chez ''Escherichia coli'' K12 avec un seuil de 0.5 pour la confiance dans une arête et '''ouvrez'''-le dans Cytoscape (menu <tt>File->Import->Network</tt>). | ||
| - | Par défaut, les sommets du graphes sont disposés sur une grille. '''Expérimentez''' différents algorithmes de dessin (layout in english donc dans le menu <tt>layout</tt>). Attention, certains ne sont pas adaptés et d'autres plutôt gourmands en temps de calcul. | + | Par défaut, les sommets du graphes sont disposés sur une grille. '''Expérimentez''' différents algorithmes de dessin (layout in english donc dans le menu <tt>layout</tt>). Attention, certains ne sont pas adaptés et d'autres plutôt gourmands en temps de calcul. |
== Attributs sur les sommets et arêtes == | == Attributs sur les sommets et arêtes == | ||
| Line 70: | Line 70: | ||
= Parcours en profondeur d'un graphe = | = Parcours en profondeur d'un graphe = | ||
| - | Il s'agit à présent d'implémenter le parcours en profondeur d'un graphe en python | + | Il s'agit à présent d'implémenter le parcours en profondeur (DFS) d'un graphe en python. |
| - | + | Pour cela, vous allez réaliser une bibliothèque pour les graphes (orientés ou non). Pour DFS, vous allez implémenter le diagramme de classes (UML) suivant : | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | [[Image:M1MABS Graphes ClassDiagram1.png]] | |
| - | + | Ceci se traduit en python par le squelette suivant : [[File:M1MABS Graphes Graph-TP1-skeleton.py]] | |
| - | + | '''Copiez''' le code fourni dans un fichier Graph.py qui correspondra à votre librairie et '''testez''' le (<tt>chmod +x Graph.py && ./Graph.py</tt>). | |
| - | + | ||
| - | + | ||
| - | + | Vous pouvez aussi affichez la documentation incluse dans le code avec la commande <tt>pydoc Graph</tt> | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | Ajoutez la méthode loadSIF() pour pouvoir charger un fichier au format SIF (simplifié) : | |
| + | <source lang='python'> | ||
| + | # OPEN FILE | ||
| + | with open(filename) as f: | ||
| + | # SKIP COLUMNS NAMES | ||
| + | tmp = f.readline() | ||
| + | # PROCESS THE REMAINING LINES | ||
| + | row = f.readline().rstrip() | ||
| + | while row: | ||
| + | vals = row.split('\t') | ||
| + | self.addEdge(vals[0], vals[2]) | ||
| + | row = f.readline().rstrip() | ||
| + | </source> | ||
| + | Pour cela, vous aurez besoin d'implémenter les méthodes <tt>addEdge</tt> et <tt>addNode</tt>. Chargez le fichier [[File:M1MABS Graphe dressing.sif]]. | ||
| - | + | '''Affichez''' l'ordre du graphe ainsi chargé ainsi que le nombre d'arcs. | |
| + | |||
| + | '''Implémenter''' ensuite le parcours en profondeur dont on vous rappelle l'algorithme : | ||
| + | |||
| + | DFS(G) | ||
| + | '''for each''' vertex u <math>\in</math> V(G) | ||
| + | '''do''' color[u] <math>\leftarrow</math> WHITE | ||
| + | pred[u] <math>\leftarrow</math> NIL | ||
| + | time <math>\leftarrow</math> 0 | ||
| + | '''for each''' vertex u <math>\in</math> V(G) | ||
| + | '''do if''' color[u] = WHITE | ||
| + | '''then''' DFS-VISIT(u) | ||
| + | DFS-VISIT(u) | ||
| + | color[u] <math>\leftarrow</math> GRAY | ||
| + | time <math>\leftarrow</math> time + 1 | ||
| + | d[u] <math>\leftarrow</math> time | ||
| + | '''for each''' vertex v <math>\in</math> Adj[u] | ||
| + | '''do if''' color[v] = WHITE | ||
| + | '''then''' pred[v] <math>\leftarrow</math> u | ||
| + | DFS-VISIT(v) | ||
| + | class[ (u,v) ] <math>\leftarrow</math> TREE EDGE | ||
| + | '''else if''' color[v] = GRAY | ||
| + | '''then''' class[ (u,v) ] <math>\leftarrow</math> BACK EDGE | ||
| + | '''else if''' d[u] > d[v] | ||
| + | '''then''' class[ (u,v) ] <math>\leftarrow</math> CROSS EDGE | ||
| + | '''else''' class[ (u,v) ] <math>\leftarrow</math> FORWARD EDGE | ||
| + | color[u] <math>\leftarrow</math> BLACK | ||
| + | time <math>\leftarrow</math> time + 1 | ||
| + | f[u] <math>\leftarrow</math> time | ||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | ''' | + | '''Implémentez''' la méthode <tt>isAcyclic()</tt> qui renvoie vrai ou faux selon que le graphe est sans circuit ou non. |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | ''' | + | '''Implémentez''' la méthode <tt>topologicalSort()</tt> qui renvoie les sommets du graphes en ayant effectué un tri topologique. |
| - | + | Autres fichiers : | |
| + | * [[Image:directed.graph.with.cycle.slide29.png|thumb|100px|directed.graph.with.cycle.slide29]] [[File:directed.graph.with.cycle.slide29.sif]] | ||
Current revision as of 08:55, 18 February 2016
Contents |
Données
Les données sur lesquelles vous allez travailler provienne de la base de données STRING v8.2. Tout d'abord, trouvez le site de STRING et visualisez les partenaires de wbbJ d'Escherichia coli K12-MG1655. Les "boutons" confidence, evidence et actions permettent de modifier le rendu du graphe d'interaction en affichant soit des arêtes de tailles reflétant le niveau de confiance du lien, soit des multi-arêtes indiquant les sources de données intervenant dans un lien, soit le type d'interaction. Le bouton more permet de récupérer davantage de protéines.
Visualisation avec Cytoscape
Cytoscape est un logiciel de visualisation et d'exploration de graphes. Il a été développé dans le cadre de graphes et réseaux biologiques. Un certain nombre de plugins ou greffons sont disponibles par exemple pour récupérer/télécharger des voies métaboliques (BioCyc) directement depuis Cytoscape.
Importation d'un fichier sif et algorithmes de dessin
Vous allez dans un premier temps utiliser un graphe au format sif (simple interaction format). Il s'agit d'un format de stockage très simple de la forme :
ABC coexpression YAEE ABC coexpression METK ABC coexpression META ACCA coexpression FABD ACCA coexpression PPA ACCB coexpression HPT ...
où coexpression est le type de l'arête.
Récupérez le fichier String_EcolA_coexpression.sif contenant une partie du réseau d'interaction basé sur la co-expression chez Escherichia coli K12 avec un seuil de 0.5 pour la confiance dans une arête et ouvrez-le dans Cytoscape (menu File->Import->Network).
Par défaut, les sommets du graphes sont disposés sur une grille. Expérimentez différents algorithmes de dessin (layout in english donc dans le menu layout). Attention, certains ne sont pas adaptés et d'autres plutôt gourmands en temps de calcul.
Attributs sur les sommets et arêtes
Il est possible de charger des attributs associés aux sommets et/ou aux arêtes. Il s'agit là encore d'un format très simple. Pour les attributs portant sur les sommets, il prend la forme suivante :
Description AAS = 2-acyl-glycerophospho-ethanolamine acyltransferase; acyl-acyl-carrier protein synthetase AAT = leucyl, phenylalanyl-tRNA-protein transferase ABC = ATP-binding component of a transporter ...
où la première ligne contient le nom de l'attribut. Vous trouverez plus d'information dans la documentation de Cytoscape.
Pour les arêtes, le format est très similaire :
coexpression ABC (coexpression) YAEE = 831 ABC (coexpression) METK = 590 ABC (coexpression) META = 663 ACCA (coexpression) FABD = 537 ACCA (coexpression) PPA = 543 ACCB (coexpression) HPT = 566 ...
Récupérez le fichier contenant les annotations des gènes - EcolA_Genes_Description.noa - et chargez-le (Menu File->Import->Node Attributes) dans Cytoscape. De même, pour les niveaux de confiance sur les liens de coexpression avec le fichier String_EcolA_coexpression.eda.
Comme dans la plupart des logiciels et systèmes d'exploitation, un clic droit sur un objet fait apparaître un menu contextuel dans Cytoscape. Un clic droit sur une arête ou un sommet fait apparaître un tel menu. Remarquez la dernière entrée du menu : LinkOut qui permet d'ouvrir une URL, typiquement à partir de l'identifiant du sommet ou de l'arête, ce qui permet par exemple d'atteindre la page de description d'une protéine dans UniProt. Il est possible d'ajouter ses propres "liens externes" à partir du menu Edit->Preferences->Properties... : pour un lien externe à partir d'un noeud, il faut ajouter une propriété nodelinkouturl.NOM_DU_LIEN, par exemple et c'est ce que vous devrez faire nodelinkouturl.CGDB. Dans le champ valeur, il s'agit de spécifier l'URL avec une ou plusieurs variables, ici nous utiliserons simple l'identifiant du noeud noté %ID% : http://www-abcdb.biotoul.fr/#/entry/findbestmatch/ID/EcolA.%ID%. Pour les liens sur les arêtes, il s'agit de la propriété edglelinkouturl.NOM_DU_LIEN avec %ID1% et %ID2% les extrémités de l'arête, cf. la documentation de Cytoscape pour plus de détails.
Rendu à partir des valeurs des attributs : VizMapper
Explorez à présent les possibilités offertes par l'outil VizMapper. Essayez notamment de faire afficher des épaisseurs d'arêtes proportionnelles à la confiance du lien de coexpression.
Sélection, filtres et opérations sur les graphes
Une boite de recherche est disponible au niveau de la barre d'outil. Essayez-la pour par exemple sélectionner le gène wbbJ. Il s'agit en fait d'un filtrage sur la sélection de tous les sommets (ou toutes les arêtes). Pour élaborer un filtre plus complexe, vous avez à gauche de la boite de recherche un bouton vous permettant d'éditer les paramètres du filtre. Sélectionnez toutes les arêtes ayant une valeur de coexpression supérieure à 800. Une fois la sélection réalisée, les boutons encore plus à gauche vous permettent d'extraire un sous graphe à partir de la sélection. Par défaut, il s'agit d'un sous graphe induit par les sommets sélectionnés. Après avoir réalisé la sélection des arêtes >800, le menu Select->Nodes->Nodes connected by selected edges vous permet de sélectionner les extrémités des arêtes sélectionnées.
Importez à présent le graphe contenu dans le fichier String_EcolA_experimental.sif correspondant aux données STRING portant sur les interactions protéine-protéine. Importez également la confiance de ces relations à partir du fichier String_EcolA_experimental.eda.
Vous pouvez fusionner les 2 graphes en les sélectionnant tous les deux (maintenir la touche Control enfoncée) puis en allant dans le menu Plugins->Advanced Network Merge. Une fois cette opération réalisée, modifiez le rendu pour avoir des épaisseurs d'arêtes proportionnelles à la coexpression et un dégradé de couleur d'arête reflétant la confiance dans le lien d'interaction protéine-protéine.
Importation d'un graphe au format table
Il est également possible d'importer un graphe dans un format tabulé, ce qui permet de charger plusieurs attributs en même temps sur les arêtes. Récupérez le fichier String_EcolA_selection.tab qui contient les deux fichiers précédents ainsi que le score combiné des différentes sources disponibles dans STRING. Le fichier est de la forme :
id1 id2 experimental coexpression combined AAS SECA 546 0 546 AAS ACPP 653 0 653 ABC YAEE 900 831 999 ABC METK 0 590 591 ABC META 0 663 663 ACCA FABD 0 537 997 ...
Parcours en profondeur d'un graphe
Il s'agit à présent d'implémenter le parcours en profondeur (DFS) d'un graphe en python.
Pour cela, vous allez réaliser une bibliothèque pour les graphes (orientés ou non). Pour DFS, vous allez implémenter le diagramme de classes (UML) suivant :

Ceci se traduit en python par le squelette suivant : File:M1MABS Graphes Graph-TP1-skeleton.py
Copiez le code fourni dans un fichier Graph.py qui correspondra à votre librairie et testez le (chmod +x Graph.py && ./Graph.py).
Vous pouvez aussi affichez la documentation incluse dans le code avec la commande pydoc Graph
Ajoutez la méthode loadSIF() pour pouvoir charger un fichier au format SIF (simplifié) :
# OPEN FILE with open(filename) as f: # SKIP COLUMNS NAMES tmp = f.readline() # PROCESS THE REMAINING LINES row = f.readline().rstrip() while row: vals = row.split('\t') self.addEdge(vals[0], vals[2]) row = f.readline().rstrip()
Pour cela, vous aurez besoin d'implémenter les méthodes addEdge et addNode. Chargez le fichier File:M1MABS Graphe dressing.sif.
Affichez l'ordre du graphe ainsi chargé ainsi que le nombre d'arcs.
Implémenter ensuite le parcours en profondeur dont on vous rappelle l'algorithme :
DFS(G) for each vertex uV(G) do color[u]
WHITE pred[u]
Implémentez la méthode isAcyclic() qui renvoie vrai ou faux selon que le graphe est sans circuit ou non.
Implémentez la méthode topologicalSort() qui renvoie les sommets du graphes en ayant effectué un tri topologique.
Autres fichiers :
- File:Directed.graph.with.cycle.slide29.sif
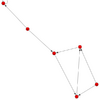 directed.graph.with.cycle.slide29
directed.graph.with.cycle.slide29