


M1 MABS BBS BGPG TD GRNs - 2015
From silico.biotoul.fr
Au cours de ce TD, nous allons utiliser une librairie (minet) faisant partie de la suite Bioconductor afin d'inférer un réseau de régulation à partir de données d'expression.
Une fois le réseau reconstruit, il s'agira d'évaluer sa qualité.
Ensuite, on pourra également visualiser le réseau obtenu avec Cytoscape par exemple.
Contents |
Principe
- Calcul d'une matrice d'information mutuelle (ou de corrélations) entre chaque paire de profils d'expression
- pour cela plusieurs estimateurs sont disponibles et utilisent, pour la plupart, des données nominales. Il faudra donc effectuer une discrétisation au préalable (fonctions fournies dans la librairie).
- Inférence du réseau
- méthodes : CLR, ARACNe, et MRNET
- Evaluation du réseau obtenu par rapport à un jeu de référence
- courbes Precision-Recall et ROC
- Choix d'un seuil, extraction du graphe obtenu et visualisation sous Cytoscape
- Partitionnement du réseau et recherche de fonctions biologiques sur-représentées au sein des clusters
Données
- Pour l'inférence : Compendium de données d'expression
L'idée est de récupérer des données d'expression depuis GEO sur l'organisme qui nous intéresse. Ici, on prendra Escherichia coli. Un certain nombre de design de microarray (GPLxxx) sont disponibles. Ceux arborant le plus d'hybridations (GSMxxx) sont GPL199 (52 séries GSExxx et 938 hybridations GSMxxx au 6 février 2013) et GPL3154 (121 séries et 956 hybridations au 6 février 2013 également).
Après avoir choisi d'utiliser GPL3154, les informations sur les 121 séries ont été étudiées pour sélectionner les plus pertinentes (en fonction de la souche dont proviennent les ARNm extraits, des conditions expérimentales, etc.). Au total, cela fait un peu plus de 800 hybridations. Le fichier GPL et les fichiers GSM ont été téléchargés. Les données ont ensuite été normalisées de manière à pouvoir comparer le niveau d'expression d'un gène dans une condition par rapport à son niveau d'expression dans une autre condition (cf. ici pour la procédure de chargement et de normalisation des données au format .CEL d'Affymetrix). Après normalisation, les données ont été prétraités afin d'avoir une matrice gènes-conditions. Pour d'associer un identifiant de spot/probeset à un gène, le fichier donnant les séquences des oligonucléotides des spots a été téléchargé depuis le site d'Affymetrix (le fabricant du microarray). Pour chaque spot, donc à partir des séquences d'oligonucléotides lui correspondant, un blast nucléique a été réalisé sur le génome afin d'identifier quelles étaient les régions complémentaires. Dans le cas où tous les oligonucléotides d'un spot ne pouvaient s'hybrider qu'avec un seul gène, l'association a été effectuée. Le résultat vous est fourni dans le fichier suivant (click droit puis enregistrer sous sinon vous allez afficher le contenu qui fait 54Mo):
- Pour la validation : Réseau de référence
Pour les données de référence, nous allons utiliser celles fournies par RegulonDb. Il s'agit donc de récupérer le réseau mis à disposition (celui entre les facteurs de transcriptions et les opérons, c'est-à-dire celui dans la partie Regulatory Network Interactions - TF - operon interactions). Pour simplifier la suite, enregistrez-le sous le nom RegulonDB--network_tf_operon.txt
Arrivés à ce stade, il nous faut identifier les gènes présents à la fois dans nos données d'expression et dans le réseau fourni par RegulonDb.
Pour cela, un prétraitement a été effectué pour associer un identifiant de gène des données d'expression à un identifiant de gène de RegulonDb :
Etudiez le script perl suivant puis utilisez-le afin d'obtenir un fichier fournissant les associations : identifiant de gène RegulonDb <-> identifiant CGDB <-> identifiant d'opéron RegulonDb. Pour le langage Perl, vous trouverez un (bref) aperçu sur la page Perl_BioPerl_recipes.
map_cgdbIds_to_RegulonDbIds.pl
La ligne commande pour générer le fichier est la suivante (le signe supérieur redirige la sortie du script dans le fichier regid_cgdbid_utid.txt qui sera créé ou remplacé s'il existait déjà) :
perl map_cgdbIds_to_RegulonDbIds.pl > regid_cgdbid_utid.txt
ou bien, si le script possède le droit d'exécution (chmod +x map_cgdbIds_to_RegulonDbIds.pl) :
./map_cgdbIds_to_RegulonDbIds.pl > regid_cgdbid_utid.txt
Visualisation du jeu de données de référence
Afin de charger le réseau de RegulonDb, il faut convertir le fichier fourni dans un format compréhensible par Cytoscape. Nous utiliserons le format SIF.
Inspirez-vous du script Perl précédent pour :
- charger le mapping RegulonDbGene -- CgdbGene -- Operon du fichier généré à l'étape précédente (regid_cgdbid_utid.txt)
- parser le fichier RegulonDB--network_tf_operon.txt et générer une arête pour chaque ligne du fichier
La sortie du script devrait ressembler à cela :
lancement du script: ./regulondb_network--2--cytoscape_sif.pl > EcolA.grn.sif début du fichier généré: head EcolA.grn.sif acrR repression acrAB acrR repression acrR ada induction alkB ada repression alkB ada induction aidB ada induction alkA adiY induction adiA adiY induction gadAXW adiY induction gadBC adiY induction gltBDF
Chargez le réseau obtenu dans Cytoscape et modifier le style d'affichage pour obtenir quelque chose comme ça :
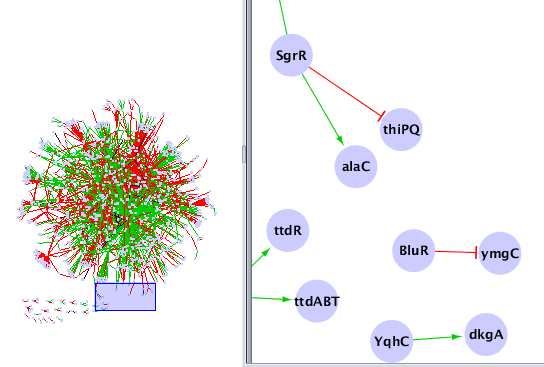
Profils d'expression par opéron
Afin de gagner du temps (de calcul) et aussi car la qualité du réseau inféré semble meilleure (Faith et al. 2007), nous allons utiliser le profil d'expression moyen par opéron et non directement celui des gènes.
Pour cela, utilisez le script average_profiles_by_regulondb_operons.pl
L'étude et la compréhension de ce script ne pourra que vous être bénéfique.
Inférence du réseau de régulation
Nous allons utiliser maintenant la librairie minet (Meyer PE, Lafitte F, Bontempi G., 2008) pour reconstruire le réseau. Une version plus complète de la vignette (nom de la documentation/tutorial en R) se trouve ici.
Librairie R
- Installer la librairie minet de Bioconductor