


Atelier Phylogénomique PorthoMCL
From silico.biotoul.fr
m (Created page with '==PorthoMCL==') |
m (→PorthoMCL) |
||
| Line 1: | Line 1: | ||
| - | ==PorthoMCL== | + | ==Liens== |
| + | * [http://silico.biotoul.fr/p/Atelier_Phylog%C3%A9nomique Atelier de Phylogénomique] | ||
| + | * [https://github.com/etabari/PorthoMCL PorthoMCL] | ||
| + | |||
| + | PorthoMCL: Parallel orthology prediction using MCL for the realm of massive genome availability | ||
| + | Ehsan Tabari and Zhengchang Su | ||
| + | Big Data Analytics 2017 2:4 [http://bdataanalytics.biomedcentral.com/articles/10.1186/s41044-016-0019-8 DOI: 10.1186/s41044-016-0019-8] | ||
| + | |||
| + | Il s'inspire d'[https://www.ncbi.nlm.nih.gov/pmc/articles/PMC403725/ orthoMCL] | ||
| + | |||
| + | <pre style="color:red;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -o-pre-wrap"> | ||
| + | Question 1.8: | ||
| + | Rappelez les différentes étapes réalisées par le logiciel. | ||
| + | Précisez les paramètres utilisés pour identifier les paires de gènes orthologues. | ||
| + | </pre> | ||
| + | ==Mise en oeuvre== | ||
| + | Le logiciel est disponible ici: /home/formation/public_html/M2_Phylogenomique/PorthoMCL-master. Il est basé sur l'enchainement de programmes ''python''. Ils peuvent être lancés séparément ou automatiquement à l'aide d'un script shell. | ||
| + | |||
| + | MSK | ||
| + | <!-- | ||
| + | <pre style="color:grey;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | mkdir -p ~/work/Prochlorococcus/PorthoMCL_auto12/0.input_faa | ||
| + | cd ~/work/Prochlorococcus/PorthoMCL_auto12 | ||
| + | cp /home/formation/public_html/M2_Phylogenomique/data/Prochlorococcus/prokka/A*/*.faa 0.input_faa/. | ||
| + | srun --nodes=12 --pty bash | ||
| + | /home/formation/public_html/M2_Phylogenomique/PorthoMCL-master/porthomcl.sh -t 12 -e 7 | ||
| + | </pre> | ||
| + | --> | ||
| + | <!-- | ||
| + | Nous allons utiliser une version plus simple: | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | mkdir ~/work/Prochlorococcus/PorthoMCL | ||
| + | cd ~/work/Prochlorococcus/PorthoMCL | ||
| + | srun --pty bash ~/work/scripts/short_PorthoMCL.sh | ||
| + | </pre> | ||
| + | <pre style="color:red;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | Question 1.9: | ||
| + | Editez le script et donnez une description des différentes tâches réalisées. | ||
| + | Que proposeriez-vous pour l'améliorer? | ||
| + | </pre> | ||
| + | --> | ||
| + | ====Step 4: Parse BLAST results==== | ||
| + | Comme les différents blastp ont déjà été réalisés, nous passons directement à l'étape 4. | ||
| + | |||
| + | Mais nous devons: | ||
| + | =====Concaténer les fichiers de résultats blastp par souche:===== | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | mkdir -p ~/work/Prochlorococcus/PorthoMCL/3.blastres | ||
| + | </pre> | ||
| + | |||
| + | MSK | ||
| + | <syntaxhighlight lang="bash"> | ||
| + | for i in ~/work/Prochlorococcus/peptide/*.faa; | ||
| + | do | ||
| + | ip=$(basename $i .faa) | ||
| + | outfile=~/work/Prochlorococcus/PorthoMCL/3.blastres/$ip.tab; | ||
| + | if [ -f $outfile ]; then | ||
| + | echo "rm $outfile" | ||
| + | rm $outfile | ||
| + | fi | ||
| + | for j in ~/work/Prochlorococcus/peptide/*.faa; | ||
| + | do | ||
| + | jp=$(basename $j .faa) | ||
| + | infile=~/work/Prochlorococcus/BlastP/$ip"_"$jp".tab" | ||
| + | if [ -f $infile ]; then | ||
| + | echo "$infile" | ||
| + | cat $infile >> $outfile | ||
| + | else | ||
| + | echo "Error $infile not found" | ||
| + | fi | ||
| + | done | ||
| + | done | ||
| + | </syntaxhighlight> | ||
| + | |||
| + | =====Modifier le nom des protéines dans les fichiers blast et fasta===== | ||
| + | Pour que les programmes suivants identifient le nom de souche et le nom de gène, il est nécessaire de rajouter un séparateur ('|') dans les noms des protéines: Aaaa.g_00001 devient Aaaa|.g_00001. Pour cela, nous pouvons utiliser la commande sed. | ||
| + | |||
| + | Dans les fichiers ~/work/Prochlorococcus/PorthoMCL/3.blastres/*.tab | ||
| + | |||
| + | MSK | ||
| + | <syntaxhighlight lang="bash"> | ||
| + | for file in ~/work/Prochlorococcus/PorthoMCL/3.blastres/*.tab | ||
| + | do | ||
| + | prefix=$(basename "$file") | ||
| + | sed -i -r 's/([A-Z][a-z]{3})(\w*\.)/\1|\2/g' $file | ||
| + | done | ||
| + | </syntaxhighlight> | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap">mkdir -p ~/work/Prochlorococcus/PorthoMCL/compliantFasta | ||
| + | </pre> | ||
| + | |||
| + | et dans les fichiers ~/work/Prochlorococcus/peptide/*.faa | ||
| + | |||
| + | MSK | ||
| + | <syntaxhighlight lang="bash"> | ||
| + | for file in ~/work/Prochlorococcus/peptide/*.faa | ||
| + | do | ||
| + | prefix=$(basename $file .pep) | ||
| + | sed -r 's/([A-Z][a-z]{3})/\1|/g' $file > ~/work/Prochlorococcus/PorthoMCL/compliantFasta/$prefix.fasta | ||
| + | done | ||
| + | </syntaxhighlight> | ||
| + | |||
| + | =====Changer le format des fichier blastp===== | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | mkdir -p ~/work/Prochlorococcus/PorthoMCL/4.splitSimSeq | ||
| + | </pre> | ||
| + | Exemple pour une souche: | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | /home/formation/public_html/M2_Phylogenomique/PorthoMCL-master/porthomclBlastParser /home/yquentin/work/Prochlorococcus/PorthoMCL/3.blastres/Aaaa.tab ~/work/Prochlorococcus/PorthoMCL/compliantFasta > ~/work/Prochlorococcus/PorthoMCL/4.splitSimSeq/Aaaa.ss.tsv | ||
| + | </pre> | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | head ~/work/Prochlorococcus/PorthoMCL/4.splitSimSeq/Aaaa.ss.tsv | ||
| + | </pre> | ||
| + | '''Format de sortie''': | ||
| + | query hit evalueMant evalueExp percentMatch | ||
| + | *evalueMant et evalueExp : mantisse (partie décimale) et exposant de la e-value | ||
| + | *percentMatch = longueur de l'alignement / longueur de la séquence la plus courte (query ou hit) *100 | ||
| + | Appliquez à toutes les souches. | ||
| + | |||
| + | MSK | ||
| + | <syntaxhighlight lang="bash"> | ||
| + | for file in ~/work/Prochlorococcus/peptide/*.faa | ||
| + | do | ||
| + | prefix=$(basename $file .faa) | ||
| + | /home/formation/public_html/M2_Phylogenomique/PorthoMCL-master/porthomclBlastParser ~/work/Prochlorococcus/PorthoMCL/3.blastres/$prefix.tab ~/work/Prochlorococcus/PorthoMCL/compliantFasta > ~/work/Prochlorococcus/PorthoMCL/4.splitSimSeq/$prefix.ss.tsv | ||
| + | done | ||
| + | </syntaxhighlight> | ||
| + | |||
| + | Vérifiez les fichiers obtenus. | ||
| + | |||
| + | ====Step 5: Finding Best Hits==== | ||
| + | Les paralogues sont identifiés et un score non normalisé leurs aient assignés. Les scores seront normalisés à l'étape 5.3 pour qu'ils soient comparables entre les génomes. | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | mkdir -p ~/work/Prochlorococcus/PorthoMCL/5.paralogTemp | ||
| + | mkdir -p ~/work/Prochlorococcus/PorthoMCL/5.besthit | ||
| + | </pre> | ||
| + | Créer un fichier avec la liste des taxon | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | ls -1 ~/work/Prochlorococcus/peptide/*faa | sed -r 's/.+([A-Z][a-z]{3}).faa/\1/' > ~/work/Prochlorococcus/PorthoMCL/taxon_list | ||
| + | </pre> | ||
| + | Test avec une souche: | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | /home/formation/public_html/M2_Phylogenomique/PorthoMCL-master/porthomclPairsBestHit.py -t ~/work/Prochlorococcus/PorthoMCL/taxon_list -s ~/work/Prochlorococcus/PorthoMCL/4.splitSimSeq -b ~/work/Prochlorococcus/PorthoMCL/5.besthit -q ~/work/Prochlorococcus/PorthoMCL/5.paralogTemp -x 1 | ||
| + | </pre> | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | head ~/work/Prochlorococcus/PorthoMCL/5.paralogTemp/Aaaa.pt.tsv | ||
| + | head ~/work/Prochlorococcus/PorthoMCL/5.besthit/Aaaa.bh.tsv | ||
| + | </pre> | ||
| + | Boucle sur les souches: | ||
| + | |||
| + | MSK | ||
| + | <syntaxhighlight lang="bash"> | ||
| + | for num in {1..12} | ||
| + | do | ||
| + | /home/formation/public_html/M2_Phylogenomique/PorthoMCL-master/porthomclPairsBestHit.py -t ~/work/Prochlorococcus/PorthoMCL/taxon_list -s ~/work/Prochlorococcus/PorthoMCL/4.splitSimSeq -b ~/work/Prochlorococcus/PorthoMCL/5.besthit -q ~/work/Prochlorococcus/PorthoMCL/5.paralogTemp -x $num | ||
| + | done | ||
| + | </syntaxhighlight> | ||
| + | |||
| + | ====Step 6: Finding Orthologs==== | ||
| + | A la sortie de cette étape nous obtenons l'ensemble des paires de gènes orthologues. Les fichiers de sortie sont concaténés. Le fichier concaténé est donnée en entrée à MCL. | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | mkdir -p ~/work/Prochlorococcus/PorthoMCL/6.orthologs | ||
| + | </pre> | ||
| + | Test avec une souche: | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | /home/formation/public_html/M2_Phylogenomique/PorthoMCL-master/porthomclPairsOrthologs.py -t ~/work/Prochlorococcus/PorthoMCL/taxon_list -b ~/work/Prochlorococcus/PorthoMCL/5.besthit -o ~/work/Prochlorococcus/PorthoMCL/6.orthologs -x 1 | ||
| + | </pre> | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | head ~/work/Prochlorococcus/PorthoMCL/6.orthologs/Aaaa.ort.tsv | ||
| + | </pre> | ||
| + | Boucle sur les souches: | ||
| + | |||
| + | MSK | ||
| + | <syntaxhighlight lang="bash"> | ||
| + | for num in {1..12} | ||
| + | do | ||
| + | /home/formation/public_html/M2_Phylogenomique/PorthoMCL-master/porthomclPairsOrthologs.py -t ~/work/Prochlorococcus/PorthoMCL/taxon_list -b ~/work/Prochlorococcus/PorthoMCL/5.besthit -o ~/work/Prochlorococcus/PorthoMCL/6.orthologs -x $num | ||
| + | done | ||
| + | </syntaxhighlight> | ||
| + | |||
| + | ====Step 7: Finding Paralogs==== | ||
| + | Les scores calculés à l'étape 5 sont normalisés. Pour la normalisation, PorthoMCL utilise la moyenne des scores des paralogues qui ont des orthologues. | ||
| + | |||
| + | Pour trouver les paralogues, nous devons extraire tous les gènes qui ont une relation orthologique dans chaque génome. Ceci peut être réalisé en utilisant un ensemble de commandes bash comme suit. | ||
| + | |||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | mkdir -p ~/work/Prochlorococcus/PorthoMCL/7.ogenes | ||
| + | cd ~/work/Prochlorococcus/PorthoMCL | ||
| + | </pre> | ||
| + | genes in the second column | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | awk -F'[|\t]' '{print $4 >> ("7.ogenes/"$3".og.tsv")}' 6.orthologs/*.ort.tsv | ||
| + | </pre> | ||
| + | genes in the first column | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | awk -F'[|\t]' '{print $2 >> ("7.ogenes/"$1".og.tsv")}' 6.orthologs/*.ort.tsv | ||
| + | </pre> | ||
| + | Avec ces fichiers (fichiers og) en main, nous pouvons normaliser les relations inparalogiques : | ||
| + | |||
| + | Le résultat de cette étape est l'ensemble des gènes paralogues avec des scores normalisés. | ||
| + | |||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | mkdir -p ~/work/Prochlorococcus/PorthoMCL/7.paralogs | ||
| + | </pre> | ||
| + | Test avec la première souche: | ||
| + | |||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | /home/formation/public_html/M2_Phylogenomique/PorthoMCL-master/porthomclPairsInParalogs.py -t ~/work/Prochlorococcus/PorthoMCL/taxon_list -q ~/work/Prochlorococcus/PorthoMCL/5.paralogTemp -o ~/work/Prochlorococcus/PorthoMCL/7.ogenes -p ~/work/Prochlorococcus/PorthoMCL/7.paralogs -x 1 | ||
| + | </pre> | ||
| + | |||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | head ~/work/Prochlorococcus/PorthoMCL/7.paralogs/Aaaa.par.tsv | ||
| + | </pre> | ||
| + | Boucle sur les souches: | ||
| + | |||
| + | MSK | ||
| + | <syntaxhighlight lang="bash"> | ||
| + | for num in {1..12} | ||
| + | do | ||
| + | /home/formation/public_html/M2_Phylogenomique/PorthoMCL-master/porthomclPairsInParalogs.py -t ~/work/Prochlorococcus/PorthoMCL/taxon_list -q ~/work/Prochlorococcus/PorthoMCL/5.paralogTemp -o ~/work/Prochlorococcus/PorthoMCL/7.ogenes -p ~/work/Prochlorococcus/PorthoMCL/7.paralogs -x $num | ||
| + | done | ||
| + | </syntaxhighlight> | ||
| + | |||
| + | ====Step 8: MCL==== | ||
| + | <pre style="color:red;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | Question 1.10: | ||
| + | Rappelez le principe de MCL et les paramètres utilisés. | ||
| + | Quel est l'effet de ces paramètres? | ||
| + | </pre> | ||
| + | C'est exactement comme la dernière étape d'OrthoMCL. Il suffit d'exécuter MCL sur les sorties du script : | ||
| + | |||
| + | Notez que -t définit le nombre de processeurs que vous devez utiliser pour MCL. | ||
| + | |||
| + | [http://bioinfo.genotoul.fr/index.php/how-to-use/?software=How_to_use_SLURM_MCL How to use SLURM MCL] | ||
| + | |||
| + | Location: /usr/local/bioinfo/src/MCL | ||
| + | |||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | srun --pty bash | ||
| + | #Load modules | ||
| + | module load bioinfo/mcl-14-137 | ||
| + | |||
| + | cat ~/work/Prochlorococcus/PorthoMCL/6.orthologs/*.tsv >> ~/work/Prochlorococcus/PorthoMCL/8.all.ort.tsv | ||
| + | |||
| + | mcl ~/work/Prochlorococcus/PorthoMCL/8.all.ort.tsv --abc -I 1.5 -t 4 -o ~/work/Prochlorococcus/PorthoMCL/8.all.ort.group | ||
| + | </pre> | ||
| + | Taille des OG: | ||
| + | |||
| + | Vous pouvez utiliser [http://www.shellunix.com/awk.html awk] avec NF qui est le nombre de champs de l'enregistrement courant. | ||
| + | |||
| + | MSK | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | awk 'BEGIN { | ||
| + | n=1; | ||
| + | printf("class\tsize\n"); | ||
| + | } | ||
| + | { | ||
| + | printf(n"\t"NF"\n"); | ||
| + | n = n+1; | ||
| + | }' ~/work/Prochlorococcus/PorthoMCL/8.all.ort.group > ~/work/Prochlorococcus/PorthoMCL/8.all.ort.group_class_size.tab | ||
| + | </pre> | ||
| + | |||
| + | Taux de paralogie (avec ''awk'' mais en vous inspirant du [http://silico.biotoul.fr/p/M1_BBS_Graphes_TP_Recherche_de_communaut%C3%A9s_dans_les_graphes TP5] de M1): | ||
| + | |||
| + | MSK | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | awk '{ | ||
| + | j=0; | ||
| + | for (i=1; i<=NF; i++) { | ||
| + | str=substr($i,1,4); | ||
| + | strlist[str]++; | ||
| + | j++; | ||
| + | } | ||
| + | if ( j > 12 ) { | ||
| + | nb=0; | ||
| + | for (str in strlist ) {nb++} | ||
| + | par=(NF-nb)/NF | ||
| + | printf("%i\t%i\t%5.3f", NF, nb, par) | ||
| + | for (str in strlist ) { | ||
| + | printf("\t%s:%s", str, strlist[str]); | ||
| + | } | ||
| + | printf("\n") | ||
| + | } | ||
| + | delete strlist; | ||
| + | }' ~/work/Prochlorococcus/PorthoMCL/8.all.ort.group | ||
| + | </pre> | ||
| + | |||
| + | <pre style="color:red;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | Que pensez-vous de la distribution des taille des OG? Quelle-est la taille maximum attendue? | ||
| + | </pre> | ||
| + | Paralogs | ||
| + | <pre style="color:grey;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap">cat 7.paralogs/*.tsv >> 8.all.par.tsv | ||
| + | |||
| + | mcl 8.all.par.tsv --abc -I 1.5 -t 4 -o 8.all.par.group | ||
| + | </pre> | ||
| + | |||
| + | ====Visualisation des OG==== | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | srun --pty bash | ||
| + | module load system/R-3.5.1 | ||
| + | </pre> | ||
| + | =====Vision globale avec genoplotR===== | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | Rscript ~/work/scripts/genoplot_OG_links.R --MCL_file=~/work/Prochlorococcus/PorthoMCL/8.all.ort.group --pdf_file=~/work/Prochlorococcus/images/8.all.ort.group_all.pdf | ||
| + | |||
| + | evince work/Prochlorococcus/images/8.all.ort.group_all.pdf | ||
| + | </pre> | ||
| + | [http://silico.biotoul.fr/enseignement/m2BBS/Phylogenomic/figures/8.all.ort.group_all.pdf 8.all.ort.group_all.pdf] | ||
| + | <pre style="color:red;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | Question 1.11: | ||
| + | Analysez la figure obtenue. | ||
| + | Comparez-là avec celle réalisée avec les blastn. | ||
| + | </pre> | ||
| + | |||
| + | =====Restreindre aux OG sans paralogues===== | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | Rscript ~/work/scripts/genoplot_OG_links.R --MCL_file=~/work/Prochlorococcus/PorthoMCL/8.all.ort.group --pdf_file=~/work/Prochlorococcus/images/8.all.ort.group_nopara.pdf --max_paralogs=0 | ||
| + | |||
| + | evince work/Prochlorococcus/images/8.all.ort.group.1.5-5.50_nopara.pdf | ||
| + | </pre> | ||
| + | [http://silico.biotoul.fr/enseignement/m2BBS/Phylogenomic/figures/8.all.ort.group_nopara.pdf 8.all.ort.group_nopara.pdf] | ||
| + | <pre style="color:red;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | Question 1.12: | ||
| + | Comparez les deux figures, avec et sans les paralogues. | ||
| + | Comment se répartissent les régions peu denses en gènes orthologues? | ||
| + | </pre> | ||
| + | |||
| + | =====Restreindre à un seul OG===== | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | Rscript ~/work/scripts/genoplot_OG_links.R --MCL_file=~/work/Prochlorococcus/PorthoMCL/8.all.ort.group --pdf_file=~/work/Prochlorococcus/images/8.all.ort.group_OG5.pdf --select_OG=5 | ||
| + | |||
| + | evince work/Prochlorococcus/images/8.all.ort.group_OG5.pdf | ||
| + | </pre> | ||
| + | [http://silico.biotoul.fr/enseignement/m2BBS/Phylogenomic/figures/8.all.ort.group_all.pdf 8.all.ort.group_OG5.pdf] | ||
| + | |||
| + | Liste des membres du groupe 5 | ||
| + | <pre> | ||
| + | Aaaa.g_00266 Aaaa.g_00937 Aaaa.g_01196 | ||
| + | Aaab.g_00239 Aaab.g_00820 Aaab.g_00825 | ||
| + | Aaac.g_01231 Aaac.g_01233 Aaac.g_02086 | ||
| + | Aaad.g_00743 Aaad.g_00747 Aaad.g_00909 | ||
| + | Aaae.g_00242 Aaae.g_00786 Aaae.g_00791 | ||
| + | Aaaf.g_00249 Aaaf.g_00782 Aaaf.g_00787 | ||
| + | Aaag.g_00262 Aaag.g_00862 Aaag.g_00867 | ||
| + | Aaah.g_00255 Aaah.g_00856 Aaah.g_01180 | ||
| + | Aaai.g_00757 Aaai.g_00761 Aaai.g_00954 | ||
| + | Aaaj.g_00252 Aaaj.g_00786 Aaaj.g_00791 | ||
| + | Aaak.g_00251 Aaak.g_00834 Aaak.g_00839 | ||
| + | Aaal.g_00941 Aaal.g_00943 Aaal.g_02214 | ||
| + | </pre> | ||
| + | Extraction du sous-graphe correspondant à ces gènes: | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | for i in Aaaa*g_00266 Aaaa\|.g_00937 Aaaa\|.g_01196 Aaab\|.g_00239 Aaab\|.g_00820 Aaab\|.g_00825 Aaac\|.g_01231 Aaac\|.g_01233 Aaac\|.g_02086 Aaad\|.g_00743 Aaad\|.g_00747 Aaad\|.g_00909 Aaae\|.g_00242 Aaae\|.g_00786 Aaae\|.g_00791 Aaaf\|.g_00249 Aaaf\|.g_00782 Aaaf\|.g_00787 Aaag\|.g_00262 Aaag\|.g_00862 Aaag\|.g_00867 Aaah\|.g_00255 Aaah\|.g_00856 Aaah\|.g_01180 Aaai\|.g_00757 Aaai\|.g_00761 Aaai\|.g_00954 Aaaj\|.g_00252 Aaaj\|.g_00786 Aaaj\|.g_00791 Aaak\|.g_00251 Aaak\|.g_00834 Aaak\|.g_00839 Aaal\|.g_00941 Aaal\|.g_00943 Aaal\|.g_02214 | ||
| + | do | ||
| + | grep $i ~/work/Prochlorococcus/PorthoMCL/8.all.ort.tsv | ||
| + | done | sort | uniq > ~/work/Prochlorococcus/PorthoMCL/G5.gr | ||
| + | </pre> | ||
| + | Tracez le graphe de ces gènes avec la librairie ''igraph'' (c.f. [http://silico.biotoul.fr/p/M1_BBS_Graphes_TP_Recherche_de_communaut%C3%A9s_dans_les_graphes TP5] de M1). | ||
| + | |||
| + | MSK | ||
| + | <syntaxhighlight lang="python"> | ||
| + | library(igraph) | ||
| + | gr <- read.graph("work/Prochlorococcus/PorthoMCL/G5.gr",format="ncol") | ||
| + | plot(gr, vertex.label.cex=0.8, vertex.size=5, vertex.shape="sphere", edge.width=E(gr)$weight, edge.color="red") | ||
| + | </syntaxhighlight> | ||
| + | [http://silico.biotoul.fr/enseignement/m2BBS/Phylogenomic/figures/graph_OG5.png graph_OG5.png] | ||
| + | <pre style="color:red;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | Question 1.13: | ||
| + | Commentez ces résultats. | ||
| + | Pouvez vous formuler des hypothèses sur l'évolution de ces gènes? | ||
| + | Comment vérifier ces hypothèses? | ||
| + | </pre> | ||
| + | |||
| + | =====Tester l'effet de l'IF sur la taille des OG===== | ||
| + | <!-- | ||
| + | Lancer MCL avec différents IF (respecter la syntaxe des noms!) | ||
| + | |||
| + | Le motif suivant est attendu: 8.all.ort.group.<IF>-5.50. | ||
| + | <pre style="color:blue;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | Rscript ~/work/scripts/MCL_partition_inflate.R --MCL_dir=~/work/Prochlorococcus/PorthoMCL --nb_strains=12 pdf_file=~/work/Prochlorococcus/images/8.all.ort.group_IF.pdf | ||
| + | |||
| + | evince ~/work/Prochlorococcus/images/8.all.ort.group_IF.pdf | ||
| + | </pre> | ||
| + | Code couleurs: | ||
| + | <pre> | ||
| + | bleu : tous les OG | ||
| + | rouge : OG sans paralogues | ||
| + | vert : OG constitués uniquement d'orthologues 1:1 | ||
| + | orange : OG avec au moins un paralogue(s) | ||
| + | </pre> | ||
| + | --> | ||
| + | |||
| + | =====Tester l'effet de l'IF sur la paralogie===== | ||
| + | <!-- | ||
| + | <pre style="color:grey;white-space: pre-wrap;white-space: -moz-pre-wrap;white-space: -pre-wrap;white-space: -o-pre-wrap"> | ||
| + | Rscript ~/work/scripts/MCL_OG_paralogy.R --MCL_file=~/work/Prochlorococcus/PorthoMCL/8.all.ort.group.1.5-5.50 --pdf_file=~/work/Prochlorococcus/images/8.all.ort.group.1.5-5.50_paralogs.pdf | ||
| + | Rscript ~/work/scripts/MCL_OG_paralogy.R --MCL_file=~/work/Prochlorococcus/PorthoMCL/8.all.ort.group.8.0-5.50 --pdf_file=~/work/Prochlorococcus/images/8.all.ort.group.8.0-5.50_paralogs.pdf | ||
| + | </pre> | ||
| + | --> | ||
| + | <!-- | ||
| + | ====old stuff2==== | ||
| + | |||
| + | read partition as list in R | ||
| + | [http://bioinfo.genotoul.fr/index.php/how-to-use/?software=How_to_use_SLURM_R ow_to_use_SLURM_R] | ||
| + | <div style="height: 150px; overflow:auto; border: thin solid blue; background: grey"> | ||
| + | <pre> | ||
| + | partition <- '/home/yquentin/work/Prochlorococcus/PorthoMCL/8.all.ort.group.5.50' | ||
| + | i <- 1 | ||
| + | con = file(partition, "r") | ||
| + | while ( TRUE ) { | ||
| + | line = readLines(con, n = 1) | ||
| + | if ( length(line) == 0 ) { | ||
| + | break | ||
| + | } | ||
| + | list[[i]] <- (strsplit(line, "\t", fixed = FALSE, perl = TRUE)) | ||
| + | #print(list[[i]]) | ||
| + | i <- i + 1 | ||
| + | } | ||
| + | close(con) | ||
| + | |||
| + | </pre> | ||
| + | </div> | ||
| + | |||
| + | <div style="height: 150px; overflow:auto; border: thin solid blue; background: grey"> | ||
| + | <pre> | ||
| + | directory <- '/home/yquentin/work/Prochlorococcus/PorthoMCL/4.splitSimSeq' | ||
| + | filenames <- list.files(directory, pattern = "*..ss.tsv*", full.names = TRUE) | ||
| + | nbfile <- length(filenames) | ||
| + | |||
| + | allintra <- NULL | ||
| + | for (i in 1:nbfile){ | ||
| + | cat(filenames[i], "\n") | ||
| + | data <- read.table(filenames[i], header=F, stringsAsFactors = FALSE) | ||
| + | intra <- subset(data, data[,1]!=data[,2]) | ||
| + | allintra <- rbind(allintra, intra) | ||
| + | } | ||
| + | nrow(allintra) | ||
| + | |||
| + | png(pdffile, width = 480, height = 480, units = "px", pointsize = 12, bg = "white") | ||
| + | hist(allintra$V5, nclass=200, xlim=c(0,100), xlab='percentMatch', col='lightblue', freq=F) | ||
| + | hist(allintra$V4, nclass=200, xlab='evalueExponent', col='lightblue', freq=F) | ||
| + | dev.off() | ||
| + | </pre> | ||
| + | </div> | ||
| + | --> | ||
Revision as of 10:13, 14 October 2021
Contents |
Liens
PorthoMCL: Parallel orthology prediction using MCL for the realm of massive genome availability Ehsan Tabari and Zhengchang Su Big Data Analytics 2017 2:4 DOI: 10.1186/s41044-016-0019-8
Il s'inspire d'orthoMCL
Question 1.8: Rappelez les différentes étapes réalisées par le logiciel. Précisez les paramètres utilisés pour identifier les paires de gènes orthologues.
Mise en oeuvre
Le logiciel est disponible ici: /home/formation/public_html/M2_Phylogenomique/PorthoMCL-master. Il est basé sur l'enchainement de programmes python. Ils peuvent être lancés séparément ou automatiquement à l'aide d'un script shell.
MSK
Step 4: Parse BLAST results
Comme les différents blastp ont déjà été réalisés, nous passons directement à l'étape 4.
Mais nous devons:
Concaténer les fichiers de résultats blastp par souche:
mkdir -p ~/work/Prochlorococcus/PorthoMCL/3.blastres
MSK
for i in ~/work/Prochlorococcus/peptide/*.faa; do ip=$(basename $i .faa) outfile=~/work/Prochlorococcus/PorthoMCL/3.blastres/$ip.tab; if [ -f $outfile ]; then echo "rm $outfile" rm $outfile fi for j in ~/work/Prochlorococcus/peptide/*.faa; do jp=$(basename $j .faa) infile=~/work/Prochlorococcus/BlastP/$ip"_"$jp".tab" if [ -f $infile ]; then echo "$infile" cat $infile >> $outfile else echo "Error $infile not found" fi done done
Modifier le nom des protéines dans les fichiers blast et fasta
Pour que les programmes suivants identifient le nom de souche et le nom de gène, il est nécessaire de rajouter un séparateur ('|') dans les noms des protéines: Aaaa.g_00001 devient Aaaa|.g_00001. Pour cela, nous pouvons utiliser la commande sed.
Dans les fichiers ~/work/Prochlorococcus/PorthoMCL/3.blastres/*.tab
MSK
for file in ~/work/Prochlorococcus/PorthoMCL/3.blastres/*.tab do prefix=$(basename "$file") sed -i -r 's/([A-Z][a-z]{3})(\w*\.)/\1|\2/g' $file done
mkdir -p ~/work/Prochlorococcus/PorthoMCL/compliantFasta
et dans les fichiers ~/work/Prochlorococcus/peptide/*.faa
MSK
for file in ~/work/Prochlorococcus/peptide/*.faa do prefix=$(basename $file .pep) sed -r 's/([A-Z][a-z]{3})/\1|/g' $file > ~/work/Prochlorococcus/PorthoMCL/compliantFasta/$prefix.fasta done
Changer le format des fichier blastp
mkdir -p ~/work/Prochlorococcus/PorthoMCL/4.splitSimSeq
Exemple pour une souche:
/home/formation/public_html/M2_Phylogenomique/PorthoMCL-master/porthomclBlastParser /home/yquentin/work/Prochlorococcus/PorthoMCL/3.blastres/Aaaa.tab ~/work/Prochlorococcus/PorthoMCL/compliantFasta > ~/work/Prochlorococcus/PorthoMCL/4.splitSimSeq/Aaaa.ss.tsv
head ~/work/Prochlorococcus/PorthoMCL/4.splitSimSeq/Aaaa.ss.tsv
Format de sortie:
query hit evalueMant evalueExp percentMatch
- evalueMant et evalueExp : mantisse (partie décimale) et exposant de la e-value
- percentMatch = longueur de l'alignement / longueur de la séquence la plus courte (query ou hit) *100
Appliquez à toutes les souches.
MSK
for file in ~/work/Prochlorococcus/peptide/*.faa do prefix=$(basename $file .faa) /home/formation/public_html/M2_Phylogenomique/PorthoMCL-master/porthomclBlastParser ~/work/Prochlorococcus/PorthoMCL/3.blastres/$prefix.tab ~/work/Prochlorococcus/PorthoMCL/compliantFasta > ~/work/Prochlorococcus/PorthoMCL/4.splitSimSeq/$prefix.ss.tsv done
Vérifiez les fichiers obtenus.
Step 5: Finding Best Hits
Les paralogues sont identifiés et un score non normalisé leurs aient assignés. Les scores seront normalisés à l'étape 5.3 pour qu'ils soient comparables entre les génomes.
mkdir -p ~/work/Prochlorococcus/PorthoMCL/5.paralogTemp mkdir -p ~/work/Prochlorococcus/PorthoMCL/5.besthit
Créer un fichier avec la liste des taxon
ls -1 ~/work/Prochlorococcus/peptide/*faa | sed -r 's/.+([A-Z][a-z]{3}).faa/\1/' > ~/work/Prochlorococcus/PorthoMCL/taxon_list
Test avec une souche:
/home/formation/public_html/M2_Phylogenomique/PorthoMCL-master/porthomclPairsBestHit.py -t ~/work/Prochlorococcus/PorthoMCL/taxon_list -s ~/work/Prochlorococcus/PorthoMCL/4.splitSimSeq -b ~/work/Prochlorococcus/PorthoMCL/5.besthit -q ~/work/Prochlorococcus/PorthoMCL/5.paralogTemp -x 1
head ~/work/Prochlorococcus/PorthoMCL/5.paralogTemp/Aaaa.pt.tsv head ~/work/Prochlorococcus/PorthoMCL/5.besthit/Aaaa.bh.tsv
Boucle sur les souches:
MSK
for num in {1..12} do /home/formation/public_html/M2_Phylogenomique/PorthoMCL-master/porthomclPairsBestHit.py -t ~/work/Prochlorococcus/PorthoMCL/taxon_list -s ~/work/Prochlorococcus/PorthoMCL/4.splitSimSeq -b ~/work/Prochlorococcus/PorthoMCL/5.besthit -q ~/work/Prochlorococcus/PorthoMCL/5.paralogTemp -x $num done
Step 6: Finding Orthologs
A la sortie de cette étape nous obtenons l'ensemble des paires de gènes orthologues. Les fichiers de sortie sont concaténés. Le fichier concaténé est donnée en entrée à MCL.
mkdir -p ~/work/Prochlorococcus/PorthoMCL/6.orthologs
Test avec une souche:
/home/formation/public_html/M2_Phylogenomique/PorthoMCL-master/porthomclPairsOrthologs.py -t ~/work/Prochlorococcus/PorthoMCL/taxon_list -b ~/work/Prochlorococcus/PorthoMCL/5.besthit -o ~/work/Prochlorococcus/PorthoMCL/6.orthologs -x 1
head ~/work/Prochlorococcus/PorthoMCL/6.orthologs/Aaaa.ort.tsv
Boucle sur les souches:
MSK
for num in {1..12} do /home/formation/public_html/M2_Phylogenomique/PorthoMCL-master/porthomclPairsOrthologs.py -t ~/work/Prochlorococcus/PorthoMCL/taxon_list -b ~/work/Prochlorococcus/PorthoMCL/5.besthit -o ~/work/Prochlorococcus/PorthoMCL/6.orthologs -x $num done
Step 7: Finding Paralogs
Les scores calculés à l'étape 5 sont normalisés. Pour la normalisation, PorthoMCL utilise la moyenne des scores des paralogues qui ont des orthologues.
Pour trouver les paralogues, nous devons extraire tous les gènes qui ont une relation orthologique dans chaque génome. Ceci peut être réalisé en utilisant un ensemble de commandes bash comme suit.
mkdir -p ~/work/Prochlorococcus/PorthoMCL/7.ogenes cd ~/work/Prochlorococcus/PorthoMCL
genes in the second column
awk -F'[|\t]' '{print $4 >> ("7.ogenes/"$3".og.tsv")}' 6.orthologs/*.ort.tsv
genes in the first column
awk -F'[|\t]' '{print $2 >> ("7.ogenes/"$1".og.tsv")}' 6.orthologs/*.ort.tsv
Avec ces fichiers (fichiers og) en main, nous pouvons normaliser les relations inparalogiques :
Le résultat de cette étape est l'ensemble des gènes paralogues avec des scores normalisés.
mkdir -p ~/work/Prochlorococcus/PorthoMCL/7.paralogs
Test avec la première souche:
/home/formation/public_html/M2_Phylogenomique/PorthoMCL-master/porthomclPairsInParalogs.py -t ~/work/Prochlorococcus/PorthoMCL/taxon_list -q ~/work/Prochlorococcus/PorthoMCL/5.paralogTemp -o ~/work/Prochlorococcus/PorthoMCL/7.ogenes -p ~/work/Prochlorococcus/PorthoMCL/7.paralogs -x 1
head ~/work/Prochlorococcus/PorthoMCL/7.paralogs/Aaaa.par.tsv
Boucle sur les souches:
MSK
for num in {1..12} do /home/formation/public_html/M2_Phylogenomique/PorthoMCL-master/porthomclPairsInParalogs.py -t ~/work/Prochlorococcus/PorthoMCL/taxon_list -q ~/work/Prochlorococcus/PorthoMCL/5.paralogTemp -o ~/work/Prochlorococcus/PorthoMCL/7.ogenes -p ~/work/Prochlorococcus/PorthoMCL/7.paralogs -x $num done
Step 8: MCL
Question 1.10: Rappelez le principe de MCL et les paramètres utilisés. Quel est l'effet de ces paramètres?
C'est exactement comme la dernière étape d'OrthoMCL. Il suffit d'exécuter MCL sur les sorties du script :
Notez que -t définit le nombre de processeurs que vous devez utiliser pour MCL.
Location: /usr/local/bioinfo/src/MCL
srun --pty bash #Load modules module load bioinfo/mcl-14-137 cat ~/work/Prochlorococcus/PorthoMCL/6.orthologs/*.tsv >> ~/work/Prochlorococcus/PorthoMCL/8.all.ort.tsv mcl ~/work/Prochlorococcus/PorthoMCL/8.all.ort.tsv --abc -I 1.5 -t 4 -o ~/work/Prochlorococcus/PorthoMCL/8.all.ort.group
Taille des OG:
Vous pouvez utiliser awk avec NF qui est le nombre de champs de l'enregistrement courant.
MSK
awk 'BEGIN {
n=1;
printf("class\tsize\n");
}
{
printf(n"\t"NF"\n");
n = n+1;
}' ~/work/Prochlorococcus/PorthoMCL/8.all.ort.group > ~/work/Prochlorococcus/PorthoMCL/8.all.ort.group_class_size.tab
Taux de paralogie (avec awk mais en vous inspirant du TP5 de M1):
MSK
awk '{
j=0;
for (i=1; i<=NF; i++) {
str=substr($i,1,4);
strlist[str]++;
j++;
}
if ( j > 12 ) {
nb=0;
for (str in strlist ) {nb++}
par=(NF-nb)/NF
printf("%i\t%i\t%5.3f", NF, nb, par)
for (str in strlist ) {
printf("\t%s:%s", str, strlist[str]);
}
printf("\n")
}
delete strlist;
}' ~/work/Prochlorococcus/PorthoMCL/8.all.ort.group
Que pensez-vous de la distribution des taille des OG? Quelle-est la taille maximum attendue?
Paralogs
cat 7.paralogs/*.tsv >> 8.all.par.tsv mcl 8.all.par.tsv --abc -I 1.5 -t 4 -o 8.all.par.group
Visualisation des OG
srun --pty bash module load system/R-3.5.1
Vision globale avec genoplotR
Rscript ~/work/scripts/genoplot_OG_links.R --MCL_file=~/work/Prochlorococcus/PorthoMCL/8.all.ort.group --pdf_file=~/work/Prochlorococcus/images/8.all.ort.group_all.pdf evince work/Prochlorococcus/images/8.all.ort.group_all.pdf
Question 1.11: Analysez la figure obtenue. Comparez-là avec celle réalisée avec les blastn.
Restreindre aux OG sans paralogues
Rscript ~/work/scripts/genoplot_OG_links.R --MCL_file=~/work/Prochlorococcus/PorthoMCL/8.all.ort.group --pdf_file=~/work/Prochlorococcus/images/8.all.ort.group_nopara.pdf --max_paralogs=0 evince work/Prochlorococcus/images/8.all.ort.group.1.5-5.50_nopara.pdf
Question 1.12: Comparez les deux figures, avec et sans les paralogues. Comment se répartissent les régions peu denses en gènes orthologues?
Restreindre à un seul OG
Rscript ~/work/scripts/genoplot_OG_links.R --MCL_file=~/work/Prochlorococcus/PorthoMCL/8.all.ort.group --pdf_file=~/work/Prochlorococcus/images/8.all.ort.group_OG5.pdf --select_OG=5 evince work/Prochlorococcus/images/8.all.ort.group_OG5.pdf
Liste des membres du groupe 5
Aaaa.g_00266 Aaaa.g_00937 Aaaa.g_01196 Aaab.g_00239 Aaab.g_00820 Aaab.g_00825 Aaac.g_01231 Aaac.g_01233 Aaac.g_02086 Aaad.g_00743 Aaad.g_00747 Aaad.g_00909 Aaae.g_00242 Aaae.g_00786 Aaae.g_00791 Aaaf.g_00249 Aaaf.g_00782 Aaaf.g_00787 Aaag.g_00262 Aaag.g_00862 Aaag.g_00867 Aaah.g_00255 Aaah.g_00856 Aaah.g_01180 Aaai.g_00757 Aaai.g_00761 Aaai.g_00954 Aaaj.g_00252 Aaaj.g_00786 Aaaj.g_00791 Aaak.g_00251 Aaak.g_00834 Aaak.g_00839 Aaal.g_00941 Aaal.g_00943 Aaal.g_02214
Extraction du sous-graphe correspondant à ces gènes:
for i in Aaaa*g_00266 Aaaa\|.g_00937 Aaaa\|.g_01196 Aaab\|.g_00239 Aaab\|.g_00820 Aaab\|.g_00825 Aaac\|.g_01231 Aaac\|.g_01233 Aaac\|.g_02086 Aaad\|.g_00743 Aaad\|.g_00747 Aaad\|.g_00909 Aaae\|.g_00242 Aaae\|.g_00786 Aaae\|.g_00791 Aaaf\|.g_00249 Aaaf\|.g_00782 Aaaf\|.g_00787 Aaag\|.g_00262 Aaag\|.g_00862 Aaag\|.g_00867 Aaah\|.g_00255 Aaah\|.g_00856 Aaah\|.g_01180 Aaai\|.g_00757 Aaai\|.g_00761 Aaai\|.g_00954 Aaaj\|.g_00252 Aaaj\|.g_00786 Aaaj\|.g_00791 Aaak\|.g_00251 Aaak\|.g_00834 Aaak\|.g_00839 Aaal\|.g_00941 Aaal\|.g_00943 Aaal\|.g_02214 do grep $i ~/work/Prochlorococcus/PorthoMCL/8.all.ort.tsv done | sort | uniq > ~/work/Prochlorococcus/PorthoMCL/G5.gr
Tracez le graphe de ces gènes avec la librairie igraph (c.f. TP5 de M1).
MSK
library(igraph) gr <- read.graph("work/Prochlorococcus/PorthoMCL/G5.gr",format="ncol") plot(gr, vertex.label.cex=0.8, vertex.size=5, vertex.shape="sphere", edge.width=E(gr)$weight, edge.color="red")
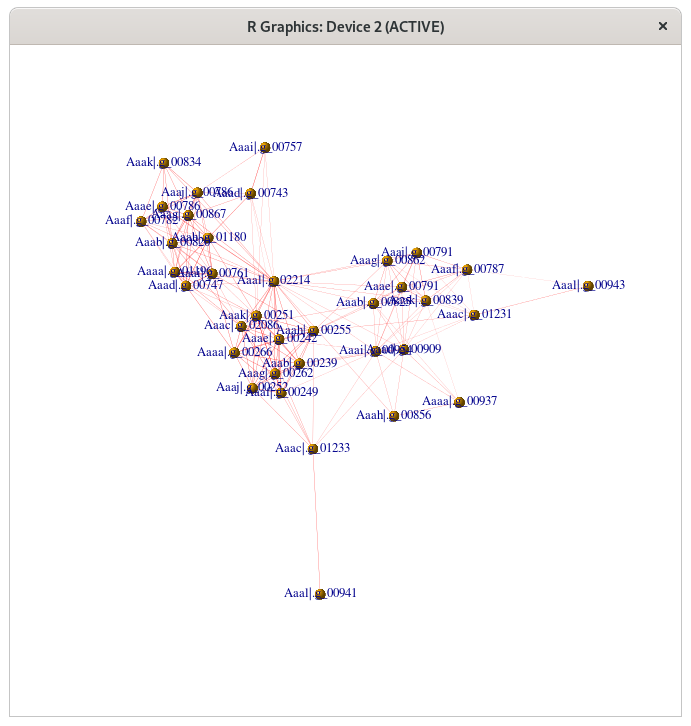{kind=link}
Question 1.13: Commentez ces résultats. Pouvez vous formuler des hypothèses sur l'évolution de ces gènes? Comment vérifier ces hypothèses?