


M2BBS - IDH
From silico.biotoul.fr
Contents |
BioMart
Premier pas
- Allez sur le site de l'EBI, puis sur BioMart pour récupérer les entrées du dataset Ensembl bacteria 14 d'Escherichia coli K-12.
- Essayer différents filtres (ayant des termes GO, ou autre).
- Sélectionner certains attributs.
- Utiliser la fonctionnalité count puis results.
Installation locale
- Aller sur le site de BioMart pour récupérer la dernière version.
- Suivre le manuel utilisateur : Quick start pour mettre à disposition un jeu de données distant depuis votre serveur.
Ajout de sources locales et édition de liens
- Télécharger le jeu de données gene (à la fin de cette page).
- Créer une base de données MySql pour héberger les tables gene et strain, avec le schéma suivant :
- gene(strain, gene, num, protein, start, end, function, uniprot)
- strain(strain, taxonomy_id, name, species)
- Alimenter ces tables avec les données téléchargées
- Ajouter le dataset gene à votre serveur BioMart et créer un Access point correspondant
- Ajouter un lien entre gene et strain
- Ajouter un lien entre gene.uniprot et le jeu de données distant unimart
Services Web
- Suivre l'exemple donné dans la documentation pour effectuer une requête sur votre instance BioMart afin de récupérer un jeu de données depuis un script python ou perl
Autres sources de données
- Répartissez-vous le reste des données disponibles en fin de page (sauf BioCyc, donc GO, profils phylogénétiques, String, et transcriptome) pour créer les tables correspondantes, faire le lien avec gene sur le type de données que vous aurez choisi, puis faire le lien avec les types de données hébergés par les instances de vos collègues.
Confrontation des données : approche ensembliste
Algorithme :
- charger les ensembles de gènes correspondant à un critère biologique
- pour chacun de ces ensembles
- calculer la probabilité d'obtenir un intersection aussi importante avec l'ensemble d'intérêt
- trier les resultats
- afficher ceux qui sont significatifs (p-valeur <= seuil ou <= seuil ajusté par la FDR)
Rappel sur la FDR:
Pour m p-valeurs calculées (m tests statistiques effectués), le seuil alpha est ajusté de la manière suivante : tout d'abord les p-valeurs sont triées de manière croissante. Ensuite, elles sont déclarées significatives tant que ce qui suit est vérifié pour k allant de 1 à m :
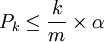
Quelques lignes de code utiles :
#!/usr/bin/perl use strict; use Data::Dumper; use Getopt::Long; use Class::Struct qw(struct); use Math::NumberCruncher; # COMMAND LINE PARAMETERS # e.g. ./search_enriched_sets.pl --sets EcolA.biocyc.sets --query 'ALAS ARGS ASNS ASPS CYSS GLTX GLYQ GLYS HISS ILES' my %params; GetOptions(\%params, 'sets=s', 'query=s', 'alpha=f', 'adjust=s', 'help' ); my $defaults = { 'alpha'=> 0.05, 'adjust' => 'y' }; sub usage { print STDERR << "EOF"; usage: $0 --sets str --query str [--alpha float] [--adjust y/n] parameters: --sets str sets file name --query str list of genes to investigate --alpha float threshold for p-value significance (default: $defaults->{alpha}) --adjust str adjust threshold for multiple testing (FDR) (default: $defaults->{adjust}) EOF } if ($params{help} || !defined($params{sets}) || !defined($params{query}) ) { usage; exit(0); } # GLOBAL VARIABLES my $sets; # reference sets to be search my $population_size; # number of possible elements (= number of genes) my %query; # query genes as keys my $alpha = defined $params{alpha} ? $params{alpha} : $defaults->{alpha}; my $adjust = defined $params{adjust} ? $params{adjust} : $defaults->{adjust}; # LOAD SETS sub load { my ($filename) = @_; open(F,$filename) || die "Cannot open $filename for input"; my $H; my $version = 1.0; my %pop; while (<F>) { ($version) = $_ =~ /^#\s*version:\s*(\S+)\s*$/ if $_ =~ /#\s*version:/; next if $_ =~ /^#/; # SKIP COMMENTS chomp; my ($id, @elements) = split /\t/; next if ! defined $id; my $desc = shift @elements if $version > 1.0; $H->{$id}->{elements} = \@elements; $H->{$id}->{description} = $desc if $version > 1.0; @pop{@elements} = @elements; } return ($H, scalar keys %pop); } struct 'ComparedSet' => [ id => '$', name => '$', common => '$', size => '$', pvalue => '$', elements => '@', commonElements => '@', ]; # MAIN # ######## # LOAD SETS ## TO DO ... # EVALUATE SETs ## TO DO ... (for each set, compute its pvalue) # PRINT SIGNIFICANT RESULTS ## TO DO ... (sort results and print them until pvalue > alpha or pvalue > adj_alpha)
Compléter le script ci-dessus afin de pouvoir analyser les ensembles suivants en utilisant le fichier File:EcolA.biocyc.sets
- ALAS ARGS ASNS ASPS CYSS GLTX GLYQ GLYS HISS ILES
- PURC PURB PURH PURE PURK PURC PURB PURH PURE
- GLPC GLPB GLPA GLPD GLPK GLPQ UGPQ GLPC GLPB GLPA GLPD
- WRBA YIEF NUOB NUOC NUOI NUOE NUOF NUOG NUON NUOA NUOL NUOK NUOJ NUOH NUOA
- HISG HISI HISA HISF HISH HISB HISC HISD PURA PURB ADK NRDB NRDA NRDD YGAD NRDE GUAB GUAA GMK PURC PURH PURE PURK PURC PURD PURL PURM PURT PYRH PYRG NDK CARA CARB PYRI PYRB PYRC PYRE PYRF PYRD PRSA PHNN DEOB HISG HISI HISA HISF HISH HISB HISC HISD PURA PURB ADK NRDB NRDA NRDD YGAD NRDE GUAB GUAA GMK PURC PURH PURE PURK PURC PURD PURL PURM PURT PYRH PYRG NDK CARA CARB PYRI PYRB
Projets 2013-14
- Barriot, R., Sherman, D., Dutour, I., How to decide which are the most pertinent overly-represented features during gene set enrichment analysis (2007) BMC Bioinformatics, 8:332
- De Preter, K., Barriot, R., Speleman, F., Vandesompele, J., Moreau, Y., Positional gene enrichment analysis of gene sets for high resolution identification of overrepresented chromosomal regions (2008) Nucleic Acids Research
Préparation des données
Il s'agit de confronter des ensembles de gène obtenus par différents critères de regroupement. Pour chacune des sources de données, il faut créer un fichier représentant les ensembles de gènes obtenus.
- Profils phylogénétiques
Les profils fournis contiennent les informations de présence d'isorthologues (Iso), d'orthologues (Ort) ou de best hit (BeH) dans d'autres souches. Il faut convertir cette matrice sous forme 0/1 pour ensuite effectuer un clustering des profils. Quelques commandes R permettent cette manipulation :
# chargement de la matrice originale m=read.table("EcolA.phylogenetic_profiles", header=T, row.names=1) # création d'une matrice de mêmes dimensions ne contenant que des 0 n=matrix(data=0, nrow=nrow(m), ncol=ncol(m), dimnames=dimnames(m)) # positionnement à 1 pour les Iso n[m=='Iso']=1 # clustering library(ade4) n.dist=dist.binary(n,1) # 1: jaccard n.dist[which(is.na(n.dist))] = 1 # si une paire de profils ne contient que des 0 hc=hclust(m, method='ave') # average linkage # sauvegarde pour utiliser un script perl pour convertir au format newick write.table(hc$merge, "EcolA.phylogenetic_profiles.01.hclust.merge", row.names=F, col.names=F) write.table(hc$labels, "EcolA.phylogenetic_profiles.01.hclust.labels", row.names=F, col.names=F, quote=F)
- Transcriptome
Utiliser MeV (installé dans /opt/) pour effectuer un clustering hiérarchique des profils d'expression fournit et enregistrer le résultat au format newick (click droit sur l'arbre, puis save as newick).
- Gene Ontology
Utiliser le script fourni (convertGO_from_categories_closure.pl), pour combiner le dag de la Gene Ontology (gene_ontology.obo) avec des associations gènes--GOTerm.
- String
Effectuer un clustering avec MCL :
- Sélectionner un score de String (coexpression ou text mining par exemple)
- Eventuellement avec un seuil pour extraire les arêtes (>=500 par exemple)
- Générer un fichier pour MCL au format une arête par ligne : gene1 gene2 score
- Vérifier que le graphe est conséquent avec Cytoscape par exemple
- Utiliser MCL et vérifier le nombre de clusters obtenus (1 par ligne)
- Reformater le fichier pour qu'il soit lisible par sookoos (ajout d'un identifiant pour les clusters) :
echo '# format: sets' > EcolA.string.mcl.sets cat -n out.EcolA.String.coexpression.edges.I20 | sed -r 's/^\s+/cluster_/' >> EcolA.string.mcl.sets
Recherche d'ensembles similaires
Utiliser le script sookoos pour analyser différentes relations, par exemple les clusters de String par rapport aux annotations de la Gene Ontology.
[scripts]$ ./sookoos Search for the most similar sets between 2 neighborhoods in terms of common elements. usage: ./sookoos [-h] [-d dissimilarity_index_type] [-a alpha_threshold] [-c multiple_adjustment_correction] -q query_file -t target_file options: -q file containing dag/tree/sets definition -t file containing dag/tree/sets definition -d dissimilarity index type (default hypergeometric), available indices : binomial, hypergeometric, ratio, mutual_coverage -a threshold for significant results (default 0.05) -c perform multiple testing adjustment (FDR) (default YES) -m maximum set size for performing comparison. Make it 0 for no limit. (default 500) -h display this help
Données et scripts
- Media:M2BBS-IDH-gene.tar.bz2
- Media:M2BBS-IDH-go.tar.bz2
- Media:M2BBS-IDH-phyogenetic_profiles.tar.bz2
- Media:M2BBS-IDH-string.tar.bz2
- Media:M2BBS-IDH-transcriptome.BsubA.tar.bz2
- Media:M2BBS-IDH-transcriptome.EcolA.tar.bz2
- Media:M2BBS-IDH-transcriptome.PaerA.tar.bz2
- Media:M2BBS-IDH-biocyc.tar.bz2