


TD4 Bioanalyse
From silico.biotoul.fr
(→Exercice 4 : Alignement multiple et construction d'une signature protéique) |
(→Exercice 4 : Alignement multiple et construction d'une signature protéique) |
||
| (25 intermediate revisions not shown) | |||
| Line 18: | Line 18: | ||
| - | ''Lors du TD3, vous avez | + | ''Lors du TD3, vous avez étudié le gène BCL2 humain. Nous allons maintenant chercher des homologues à ce gène, construire un alignement multiple et une signature protéique.'' |
| - | = Exercice 1 : recherche d' | + | = Exercice 1 : recherche d'homologues avec BlastN= |
'''1/''' A partir de la séquence de l'ARNm de l'isoforme 1 (NM_000633), lancer un BLASTN contre la banque nr (cochez la case Exclude models XM/XP)<br> | '''1/''' A partir de la séquence de l'ARNm de l'isoforme 1 (NM_000633), lancer un BLASTN contre la banque nr (cochez la case Exclude models XM/XP)<br> | ||
| + | |||
'''2/''' Regardez le résultat du BLAST et répondez aux questions suivantes : | '''2/''' Regardez le résultat du BLAST et répondez aux questions suivantes : | ||
*pourquoi y a-t-il des lettres en minuscules dans le premier alignement ? | *pourquoi y a-t-il des lettres en minuscules dans le premier alignement ? | ||
| Line 49: | Line 50: | ||
'''4/''' Récupérer les séquences en cliquant sur GenPept puis FASTA text.<br> | '''4/''' Récupérer les séquences en cliquant sur GenPept puis FASTA text.<br> | ||
| - | '''5/''' Copier les séquences dans un editeur de texte et renommer les pour avoir des noms courts : changer le nom dans l'entête FASTA, en gardant nom de protéine et de l'organisme, et sans espace (mais des tirets - ou _) <br> | + | '''5/''' Copier les séquences dans un editeur de texte et '''renommer les pour avoir des noms courts''' : changer le nom dans l'entête FASTA, en gardant nom de protéine et de l'organisme, et sans espace (mais des tirets - ou _) <br> |
Vous pouvez garder le nom fourni dans SwissProt. Par exemple : <br> | Vous pouvez garder le nom fourni dans SwissProt. Par exemple : <br> | ||
| Line 59: | Line 60: | ||
= Exercice 3 : recherche d'homologues chez le nématode (''Caenorhabditis'')= | = Exercice 3 : recherche d'homologues chez le nématode (''Caenorhabditis'')= | ||
| - | Le nématode étant assez | + | Le nématode étant assez éloigné de l'homme il n'y avait probablement pas de séquences dans le BLASTP précédent. <br> |
Vous allez maintenant utiliser le programme '''PsiBLAST''' pour chercher des homologues chez cet organisme | Vous allez maintenant utiliser le programme '''PsiBLAST''' pour chercher des homologues chez cet organisme | ||
'''1/''' A partir de la protéine précédente, sélectionner la '''banque nr''' cette fois, préciser l'organisme Caenorhabditis et choisir PsiBLAST dans la sélection des programmes de BLASTP. <br> | '''1/''' A partir de la protéine précédente, sélectionner la '''banque nr''' cette fois, préciser l'organisme Caenorhabditis et choisir PsiBLAST dans la sélection des programmes de BLASTP. <br> | ||
| - | '''2/''' Sélectionner les séquences qui s'alignent sur la zone du domaine BCL2-like et lancer la 2e itération <br> | + | '''2/''' Sélectionner les séquences qui s'alignent sur la zone du domaine BCL2-like (cocher ou décocher les cases à droite) et lancer la 2e itération <br> |
'''3/''' Observer les changements dans les résultats. Sélectionner les premières séquences, toujours alignées sur le domaine BCL2-like. Ne prenez pas les séquences de PDB (numéro d'accession commence par un chiffre). Si vous le souhaitez, lancer une seconde itération. <br> | '''3/''' Observer les changements dans les résultats. Sélectionner les premières séquences, toujours alignées sur le domaine BCL2-like. Ne prenez pas les séquences de PDB (numéro d'accession commence par un chiffre). Si vous le souhaitez, lancer une seconde itération. <br> | ||
| Line 72: | Line 73: | ||
= Exercice 4 : Alignement multiple et construction d'une signature protéique = | = Exercice 4 : Alignement multiple et construction d'une signature protéique = | ||
| - | '''1/''' Sur le site de l'EBI utiliser [http://www.ebi.ac.uk/Tools/msa/mafft/ '''MAFFT'''] pour construire un alignement multiple ( | + | '''1/''' Sur le site de l'EBI utiliser [http://www.ebi.ac.uk/Tools/msa/mafft/ '''MAFFT'''] pour construire un alignement multiple (choisir Output format : ClustalW) : regarder l'alignement, et garder cette page ouverte ! <br> |
| - | '''2/''' Visualiser l'alignement soit avec Jalview (dans | + | '''2/''' Visualiser l'alignement (ongler Result Viewers) soit avec Jalview (sur les PC, dans Programmes) soit avec Mview (en ligne) : regarder l'alignement. Jalview ou Mview sont juste des interfaces de visualisation. Où sont les parties conservées ? Voyez-vous apparaitre des groupes de séquences ? <br> |
| - | '''3/''' Copier le même alignement dans [http://weblogo.berkeley.edu/logo.cgi '''WebLogo'''] : modifier le paramètre '''Logo range''' pour cibler la zone conservée (voir selon votre alignement) et ''Logo Size per Line'' : 40 x 5 cm <br> | + | '''3/''' Copier le même alignement (Onglet Result Summary => fichier output) dans [http://weblogo.berkeley.edu/logo.cgi '''WebLogo'''] : modifier le paramètre '''Logo range''' pour cibler la zone conservée (voir selon votre alignement) et '''''Logo Size per Line''''' : 40 x 5 cm <br> |
'''4/''' Construire une signature PROSITE à partir de votre alignement (n'utilisez pas le LOGO, sinon vous ne voyez pas précisément les zones de gap) | '''4/''' Construire une signature PROSITE à partir de votre alignement (n'utilisez pas le LOGO, sinon vous ne voyez pas précisément les zones de gap) | ||
| Line 90: | Line 91: | ||
'''5/''' Tester votre signature sur [http://prosite.expasy.org/scanprosite/ '''ScanProsite'''] (choisir l'option 2) contre SwissProt ou trEMBL (plus long !) : les séquences obtenues appartiennent-elles à la famille des BCL2 ou BCL2-like ? Retrouvez-vous les mêmes organismes que précédemment ? en avez-vous d'autres ? <br> | '''5/''' Tester votre signature sur [http://prosite.expasy.org/scanprosite/ '''ScanProsite'''] (choisir l'option 2) contre SwissProt ou trEMBL (plus long !) : les séquences obtenues appartiennent-elles à la famille des BCL2 ou BCL2-like ? Retrouvez-vous les mêmes organismes que précédemment ? en avez-vous d'autres ? <br> | ||
| - | '''6/''' Regarder, par exemple dans le premier lien UniProt, comment est caractérisé ce domaine dans les banques : | + | '''6/''' Regarder, par exemple dans le premier lien UniProt, comment est caractérisé ce domaine dans les banques : dans la partie '''Family and domain databases''' cliquez sur '''''View potein in PROSITE''''' : est-ce-que ce sont plutôt des signatures ou des profiles ? <br> |
| + | ''On retiendra que les signatures (pattern en anglais) sont plutôt utilisées pour les motifs (régions assez courtes et bien conservées) et les profiles (matrices PSSM) pour des domaines protéiques'' | ||
| - | '''7/''' Pour finir, vous pouvez générer un arbre phylogénétique de vos séquences : aller sur sur le site [http://www.phylogeny.fr/ '''Phylogeny'''] dans | + | '''7/''' Pour finir, vous pouvez générer un arbre phylogénétique de vos séquences : aller sur sur le site [http://www.phylogeny.fr/ '''Phylogeny'''] dans '''''Phylogeny Analysis''''' => "Advanced" : décocher la case Multiple alignment et celle Gblocks, et cliquer sur Create workflow. Coller votre '''alignement multiple'''. Vous pouvez aussi utiliser le mode "One click" et mettre directement les séquences non alignées en format Fasta (là aussi décocher Gblocks).<br> |
| + | Essayer de comprendre l'histoire de cette famille et de voir les nœuds de spéciation et de duplication.<br> | ||
= Annexes = | = Annexes = | ||
Revision as of 11:29, 11 October 2019
Contents |
OBJECTIFS
- Comprendre le résultat du programme BLAST - Utiliser un programme d'alignement multiple, et identifier des zones conservées, générer un Logo - Ecrire une signature protéique (pattern) - Rechercher dans une banque protéique des séquences qui possèdent cette signature
Ci-dessous une sélection des sites Internet qui vous seront nécessaires :
- EBI European Bioinformatics Institute (EMBL, GB)
- NCBI National Center for Biotechnology Information (NIH, USA)
- Expasy Expert Protein Analysis System (Swiss Institute of Bioinformatics, Suisse)
- PRABI Pôle Rhône Alpes de Bio-Informatique (CNRS, Lyon)
- Phylogeny
- Génopôle Bioinformatique Toulouse
Lors du TD3, vous avez étudié le gène BCL2 humain. Nous allons maintenant chercher des homologues à ce gène, construire un alignement multiple et une signature protéique.
Exercice 1 : recherche d'homologues avec BlastN
1/ A partir de la séquence de l'ARNm de l'isoforme 1 (NM_000633), lancer un BLASTN contre la banque nr (cochez la case Exclude models XM/XP)
2/ Regardez le résultat du BLAST et répondez aux questions suivantes :
- pourquoi y a-t-il des lettres en minuscules dans le premier alignement ?
- combien d'exons composent le gène ?
- pourquoi la majorité des alignements sont sur la région 5' ?
- existe-t-il le gène BCL2 chez le poulet ?
3/ Relancer un BLASTN en précisant l'organisme que vous cherchez : quel est le résultat ?
4/ Des alignements avec des E-value >1 vous semblent-ils être de bons alignements ?
En fait, on travaillera plutôt au niveau protéique pour chercher des homologues, pour les raisons déjà vues au TP2 (conservation uniquement des CDS, pas sur les UTR, pas de problème de variation d'usage des codons donc taux d'identité plus élevé, et notion de similarité des acides aminés)
Exercice 2 : recherche de protéines homologues à BCL2 humaine
1/ Récupérer la protéine codée par l'ARNm NM_000633, et utiliser maintenant BLASTP, contre la banque SwissProt
2/ Regardez l'alignement avec la séquence BCL2-like protein 1 de poulet Q07816
- quelle est la taille de cette séquence ?
- à quoi correspond le % positives ?
- combien y a-t-il d'événement d'insertion-délétion ?
3/ Sélectionner (en cochant les cases dans le tableau) les séquences qui vous semblent homologues à notre séquence (prenez une vingtaine de séquences, a priori correspondant aux lignes rouges ou roses)
4/ Récupérer les séquences en cliquant sur GenPept puis FASTA text.
5/ Copier les séquences dans un editeur de texte et renommer les pour avoir des noms courts : changer le nom dans l'entête FASTA, en gardant nom de protéine et de l'organisme, et sans espace (mais des tirets - ou _)
Vous pouvez garder le nom fourni dans SwissProt. Par exemple :
>gi|231632|sp|P10415.2|BCL2_HUMAN RecName: Full=Apoptosis regulator Bcl-2
devient :
>BCL2_HUMAN
Exercice 3 : recherche d'homologues chez le nématode (Caenorhabditis)
Le nématode étant assez éloigné de l'homme il n'y avait probablement pas de séquences dans le BLASTP précédent.
Vous allez maintenant utiliser le programme PsiBLAST pour chercher des homologues chez cet organisme
1/ A partir de la protéine précédente, sélectionner la banque nr cette fois, préciser l'organisme Caenorhabditis et choisir PsiBLAST dans la sélection des programmes de BLASTP.
2/ Sélectionner les séquences qui s'alignent sur la zone du domaine BCL2-like (cocher ou décocher les cases à droite) et lancer la 2e itération
3/ Observer les changements dans les résultats. Sélectionner les premières séquences, toujours alignées sur le domaine BCL2-like. Ne prenez pas les séquences de PDB (numéro d'accession commence par un chiffre). Si vous le souhaitez, lancer une seconde itération.
4/ Rajouter ces quelques séquences à votre jeu de séquences précédent, en les renommant également.
Exercice 4 : Alignement multiple et construction d'une signature protéique
1/ Sur le site de l'EBI utiliser MAFFT pour construire un alignement multiple (choisir Output format : ClustalW) : regarder l'alignement, et garder cette page ouverte !
2/ Visualiser l'alignement (ongler Result Viewers) soit avec Jalview (sur les PC, dans Programmes) soit avec Mview (en ligne) : regarder l'alignement. Jalview ou Mview sont juste des interfaces de visualisation. Où sont les parties conservées ? Voyez-vous apparaitre des groupes de séquences ?
3/ Copier le même alignement (Onglet Result Summary => fichier output) dans WebLogo : modifier le paramètre Logo range pour cibler la zone conservée (voir selon votre alignement) et Logo Size per Line : 40 x 5 cm
4/ Construire une signature PROSITE à partir de votre alignement (n'utilisez pas le LOGO, sinon vous ne voyez pas précisément les zones de gap)
Pour vous aider, voici la début d'une signature (ou pattern) : Q-L-x(3)-P-x(6)-[FY]-x(2)-V-x(3)-[LVF]-[FGD]-x(2,8)-[GPS]
Vous avez probablement quelque chose d'approchant dans votre alignement, traduisant que :
Q-L : il n'y a que les acides aminés Q puis L dans 2 colonnes successives de l'alignement
x(3) : 3 colonnes avec des acides aminés variables
[FY] : dans cette colonne seuls les acides aminés F ou Y sont présents
x(2,8) : zone avec un gap. Entre 2 et 8 résidus (acides aminés) quelconques, suivant les séquences
5/ Tester votre signature sur ScanProsite (choisir l'option 2) contre SwissProt ou trEMBL (plus long !) : les séquences obtenues appartiennent-elles à la famille des BCL2 ou BCL2-like ? Retrouvez-vous les mêmes organismes que précédemment ? en avez-vous d'autres ?
6/ Regarder, par exemple dans le premier lien UniProt, comment est caractérisé ce domaine dans les banques : dans la partie Family and domain databases cliquez sur View potein in PROSITE : est-ce-que ce sont plutôt des signatures ou des profiles ?
On retiendra que les signatures (pattern en anglais) sont plutôt utilisées pour les motifs (régions assez courtes et bien conservées) et les profiles (matrices PSSM) pour des domaines protéiques
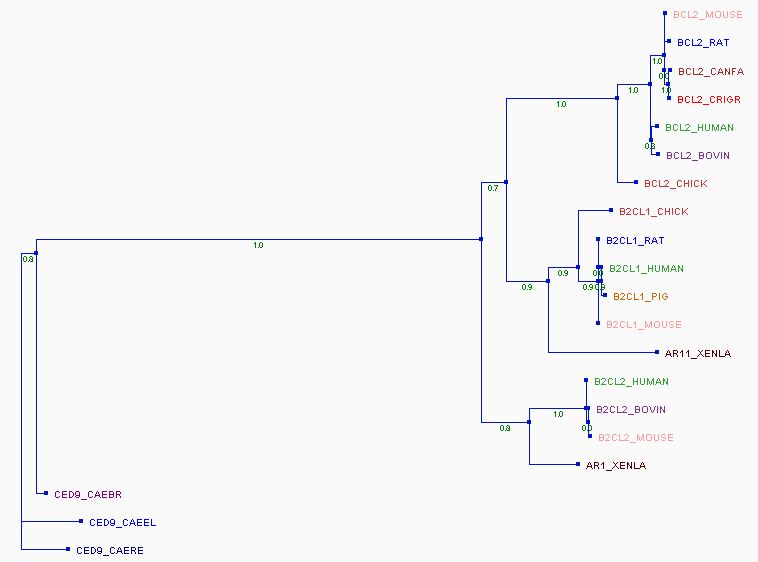
7/ Pour finir, vous pouvez générer un arbre phylogénétique de vos séquences : aller sur sur le site Phylogeny dans Phylogeny Analysis => "Advanced" : décocher la case Multiple alignment et celle Gblocks, et cliquer sur Create workflow. Coller votre alignement multiple. Vous pouvez aussi utiliser le mode "One click" et mettre directement les séquences non alignées en format Fasta (là aussi décocher Gblocks).
Essayer de comprendre l'histoire de cette famille et de voir les nœuds de spéciation et de duplication.
Annexes
Résultats du Blast P
Jeu de Séquences
ScanProsite contre TrEMBL
Arbre phylogénétique
{kind=link}