


M1 MABS BBS Math TD Proba
From silico.biotoul.fr
Contents |
Recherche de sites de fixation au ribosome
Rappel : Théorème de Bayes
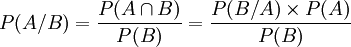 (pour plus de détails : wikipédia)
(pour plus de détails : wikipédia)
Il s'agit dans la suite de détecter la présence de RBS (Ribosome Binding Site) sur une séquence génomique :
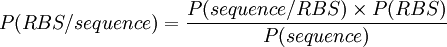
Pour se convaincre de la présence d'un motif conservé, les séquences en amont du site d’initiation de la traduction d'une centaine de séquences de Bacillus subtilis sont fournies (Séquences au format fasta). Elles sont alignées sur le codon start.
A l'aide du site WebLogo, établir le Weblogo correspondant aux séquences de B. subtilis. Où se situent les RBS ?
Calcul de P(RBS)
Pour la probabilité de présence d'un RBS, il est possible de l'approximer par le nombre de gènes (4,177) B. subtilis divisé par la taille de son génome (4,215,606 bp).
Calcul de P(sequence)
Pour la probabilité d'observer une séquence, on pourra utiliser le produit des fréquences de chaque nucléotide dans le génome Media:BsubA.fas.gz. Après avoir récupéré et décompressé le fichier, pour lire le fichier FASTA, utilisez la librairie seqinr qui est normalement installée :
library(seqinr) # chargement genome=summary(read.fasta('BsubA.fas')$BsubA01) summary(genome) # Length Class Mode #length 1 -none- numeric #composition 4 table numeric #GC 1 -none- numeric genome$length #[1] 4215606 genome$composition # # a c g t #1188073 919284 915112 1193137
Vous pouvez donc calculer f_nt, la fréquence de chaque nucléotide dans le génome et écrire une fonction qui calcule la probabilité d'observer une séquence (on suppose l'indépendance à chaque position, ce qui est complètement faux) :
p_seq = function(s, freq_res ) { # s: sequence à utiliser sous la forme de vecteur de characteres, freq_res: frequence des residus # TO DO } # test : probabilité d'observer la séquence GGAGG p_seq(c('g','g','a','g','g'), f_nt) # 0.0006258065
Calcul de P(sequence / RBS)
L'alignement des séquences de RBS de B. subtilis vous est fournit au format FASTA. Il va vous servir à calculer P(sequence/RBS). Pour cela, vous devrez construire la matrice de fréquence de chaque résidu à chaque position (la fréquence observée sert à estimer la probabilité).
known_rbs = read.fasta('Maths_Proba_Alignement_RBS.fasta') # affichage known_rbs # premiere sequence (objet sequence) known_rbs[1] # premiere sequence (sequence en nucleotides) attr(known_rbs[1], 'name') # identifiant getSequence(known_rbs[1]) # objet liste getSequence(known_rbs[1])[[1]] # 1ere sequence sous forme vecteur de caracteres
Manipulez l'objet known_rbs afin d'avoir une matrice alignment comme suit :
> head(alignment) [,1] [,2] [,3] [,4] [,5] [1,] "g" "g" "a" "g" "g" [2,] "g" "g" "a" "g" "g" [3,] "a" "g" "a" "g" "g" [4,] "g" "g" "g" "t" "g" [5,] "g" "a" "g" "g" "t" [6,] "a" "g" "g" "t" "g"
A partir de cette matrice, calculer la matrice f_res_pos qui donne la fréquence d'un résidu à une position. Vous devriez obtenir :
> round(f_res_pos,2) [,1] [,2] [,3] [,4] [,5] a 0.34 0.11 0.39 0.42 0.06 c 0.00 0.00 0.00 0.00 0.00 g 0.65 0.89 0.60 0.55 0.88 t 0.01 0.00 0.01 0.03 0.06
Vous pouvez à présent définir une fonction qui calcule P(sequence/RBS) : la probabilité d'une séquence de 5 nucléotides sachant qu'elle correspond à un RBS (produit des probabilités des résidus observés à chaque position).
Rappel : pour définir une fonction :
p_seq_rbs = function(s, freq_res_pos) { # s: sequence à utiliser sous la forme de vecteur de characteres, freq_res_pos: frequence des residus à chaque position # TO DO } # test de la fonction: probabilité d'observer la sequence GGAGG sachant que c'est un RBS p_seq_rbs(c('g','g','a','g','g'), f_res_pos) # 0.1117562
Calcul de P(RBS / sequence)
Vous pouvez maintenant écrire une fonction qui calcule 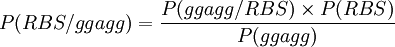 :
:
p_rbs_seq = function(s, f_res_pos, f_res, p_RBS) p_seq_rbs(s, f_res_pos) * p_RBS / p_seq(s, f_res) # test avec GGAGG p_rbs_seq(c('g','g','a','g','g'), freq_res_pos, f_nt, p_RBS) # 0.1769441
Recherche de RBS sur une séquence
Séquence test
Ecrire une fonction qui calcule la probabilité de présence d'un RBS à chaque position d'une séquence donnée. Appliquée à la séquence test, on obtient le résultat suivant :
# chargement de la séquence test newseq=as.vector(t(read.table('sequence_test.txt',colClasses='character')[1,])) newseq # recherche de RBS probas = search_rbs(newseq) probas # affichage plot(1:length(probas), probas, type="b", xaxt='n', xlab='sequence', ylab='P(RBS/sequence)') axis(1, at=1:length(probas), labels=newseq[1:length(probas)])

Contenu en information
D'après la théorie de l'information, la valeur de l'information I à la position j d'un signal est :
I(j) = ΣFijlog2Fij − ΣPilog2Pi avec Fij la fréquence du résidu i à la position j, et Pi la probabilité/fréquence théorique du résidu i.
Calculer la matrice :
I
Pseudo-comptage
Lors du calcul des fréquences de chaque résidu à chaque position dans l'alignement de départ, on observe que certaine fréquences sont nulles. Cela a pour effet d'attribuer la probabilité 0 à une position dès lors qu'un seul résidu a la malchance de tomber sur une de ces fréquences nulles.
Contenu de f_res_pos :
f_res_pos
[,1] [,2] [,3] [,4] [,5]
a 0.336538462 0.1057692 0.394230769 0.42307692 0.05769231
c 0.000000000 0.0000000 0.000000000 0.00000000 0.00000000
g 0.653846154 0.8942308 0.596153846 0.54807692 0.88461538
t 0.009615385 0.0000000 0.009615385 0.02884615 0.05769231
On obtient donc une probabilité nulle, si la séquence contient un C (quelle que soit la position) ou un T en deuxième position.
Pour éviter cela, on utilise la méthode de pseudo-comptage qui consiste à attribuer une faible probabilité plutôt que 0. Ceci est obtenu en ajoutant une faible quantité à toutes les évènements observés. La (pseudo-)fréquence est donc freq = (nboccurrences + 1) / (nbelements + 1)
Refaire donc le calcul pour obtenir le nombre d’occurrences à chaque position +1 :
[,1] [,2] [,3] [,4] [,5] a 36 12 42 45 7 c 1 1 1 1 1 g 69 94 63 58 93 t 2 1 2 4 7
Et donc les fréquences suivantes :
round( pf_res_pos ,2) [,1] [,2] [,3] [,4] [,5] a 0.33 0.11 0.39 0.42 0.06 c 0.01 0.01 0.01 0.01 0.01 g 0.64 0.87 0.58 0.54 0.86 t 0.02 0.01 0.02 0.04 0.06
Seuil pour la prédiction
A l'aide de la fonction apply, calculer les probabilités obtenues sur le jeu de séquences d'apprentissage (l'alignement). Vous devriez obtenir ceci :
> summary(true_pos_probas) Min. 1st Qu. Median Mean 3rd Qu. Max. 0.001209 0.043780 0.162500 0.140900 0.189800 0.355000
Soit un seuil de 0.001209 pour détecter tous les RBS avérés. Malheureusement, ce seuil relativement bas induit des faux positifs sur la séquence tests :
> threshold = min(true_pos_probas) > which(probas2 >= threshold) [1] 16 18 19 20 21 22 plot(1:length(probas2), probas2, type="l", xaxt='n', xlab='sequence', ylab='P(RBS/sequence)') axis(1, at=1:length(probas2), labels=newseq[1:length(probas2)]) positives = which(probas2 >= threshold) points(positives, probas2[positives] , pch=16, col='green')

Intervalle de confiance sur la moyenne de la probabilité d'un RBS dans le jeu d'apprentissage :
# INTERVALLE DE CONFIANCE A 95% m = mean(true_pos_probas) # 0.140906 abline(h=m, lwd=3) s = sd(true_pos_probas) # 0.1069476 error = qnorm(0.975)*s/sqrt(nrow(alignment)) error abline(h=m-error, lwd=2)

Attention, un tel seuil engendrera de nombre faux négatifs :
> which(true_pos_probas<m-error) [1] 3 4 5 6 8 12 13 14 17 18 21 24 26 27 28 29 31 32 34 35 36 40 41 44 45 46 47 49 50 53 58 59 60 61 63 [36] 72 75 76 77 78 81 82 84 86 93 94 97 103