


M1 Traitement de Donnees Biologiques - TP 2 R
From silico.biotoul.fr
Contents |
Régression linéaire, probabilités et intervalles de confiance
Ce TP comporte trois parties:
- Régression linéaire
- Calculs simples de probabilité
- Estimation d'un intervalle de confiance d'un paramètre (moyenne, proportion)
Créer un répertoire de travail sur le bureau (par exemple TDB-TP2) et commencez par télécharger le fichier source que vous allez utiliser et compléter pour générer le compte rendu de TP : M1.TDB.TP_reglin_proba_ic_R.Rmd (click droit de la souris -- enregistrer la cible sous...). Ouvrez le logiciel RStudio et chargez ce fichier puis lancez sa compilation pour voir le compte rendu. Pour cela cliquez sur le bouton Knit HTML ou bien utilisez la combinaison de touches Ctrl + shift + K.
Régression linéaire
On cherche à détecter une corrélation entre deux variables quantitatives mesurées sur les mêmes individus. Notre exemple: pour différentes espèces de bactéries, nous connaissons (i) la taille du génome grâce au séquençage, (ii) le nombre de séquences codantes prédites par un algorithme bioinformatique. Téléchargez et sauvegardez le fichier bacterial_genomes.txt dans le répertoire crée pour ce TP (click droit de la souris -- enregistrer la cible sous...).
Quelle(s) question(s) peut-on se poser ?
Lecture et exploration du tableau de données
Lecture:
genomes=read.table("bacterial_genomes.txt", sep="\t", header=TRUE)
Accéder directement aux colonnes du data.frame genomes simplement à partir de leurs noms (fonction attach):
attach(genomes) names(genomes) Genome_size ORF_number
Quelle est l'unité de mesure pour Genome_size ?
Faire un graphique:
plot(Genome_size,ORF_number,pch=16)
Quantifier la relation entre ces 2 variables:
- covariance
cov(Genome_size,ORF_number)
- coefficient de corrélation r de Pearson
cor(Genome_size,ORF_number)
Régression
Calculer une fonction linéaire qui relie les 2 variables, avec la commande lm() :
lm(ORF_number ~ Genome_size) # remarque: c'est une régression de "y" sur "x", d'où lm(y~x)
Gardons en mémoire le résultat de la régression:
reglin=lm(ORF_number ~ Genome_size)
On peut vérifier la significativité des coefficients de la droite avec:
summary(reglin)
Quelle est l'équation de la droite de régression ?
Calculez le coefficient de détermination R2 (% de variance expliquée par le modèle linéaire ==> bien si > 70%):
cor(Genome_size,ORF_number)^2 #(stocké aussi dans summary(reglin))
Représenter le nuage de points avec la droite de régression:
plot(Genome_size,ORF_number,pch=16) abline(reglin,col="red",lwd=2)
Ajoutez ces parties à votre compte rendu.
Calculs de probabilité
Calculs basés sur des données
L'exemple porte sur la taille des arbres dans une forêt de séquoias. Téléchargez et sauvegardez le fichier sequoia.txt (click droit de la souris -- enregistrer la cible sous...).
Chargez le fichier avec la commande read.table dans une variable sequoia, puis utilisez la commande attach pour accéder directement aux noms des colonnes. Utilisez également la commande names pour connaître le nom des colonnes dans le fichier.
Représentation graphique des données:
hist(taille_arbre)
Quelle est la probabilité qu'un arbre mesure 80m ?
length(taille_arbre[taille_arbre==80])/length(taille_arbre)
ou mieux :
length( which(taille_arbre==80) ) / length(taille_arbre)
Quelle est la probabilité qu'un arbre mesure plus de 100 ?
length( which(taille_arbre>100) ) / length(taille_arbre)
Calculs basés sur des lois (ou distributions) de probabilité
Différentes lois sont programmées (binomiale, Poisson, normale, chi2 ou χ2, ...) Il y a plusieurs fonctions pour chaque loi. Par exemple, pour la loi normale:
- dnorm() : fonction densité (density)
- pnorm() : fonction de répartition (probability)
- qnorm() : fonction quantile (quantile)
- rnorm() : générateur aléatoire (random)
Calculs basés sur une loi normale centrée réduite N(0,1)
Rappels :
- Une loi normale est définie par 2 paramètres : la moyenne et l'écart-type.
- La loi normale centrée réduite est la loi normale dont la moyenne est 0 et l'écart-type 1. Elle est notée généralement N(0,1) (0 pour la moyenne et 1 pour l'écart-type de la variable X).

Probabilité de X = 0, notée P(X = 0)
dnorm(0,0,1)
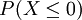 :
:
pnorm(0,0,1)
P(X > 0) :
1-pnorm(0,0,1) # ou pnorm(0,0,1, lower.tail=F)
Histogramme pour 100 valeurs échantillonnées aléatoirement suivant une N(0,1) :
hist(rnorm(100,0,1))
Application à la forêt de séquoias
Utilisez la moyenne et l'écart-type des tailles des séquoias chargées précédemment pour calculer la probabilité qu'un arbre mesure 80m selon la loi normale paramétrée ainsi.
Quelle est la probabilité qu'un séquoia mesure plus de 110m ?
Calculs basés sur une loi Binomiale B(n,p)
Exemple: pour un lot de n=100 graines d'Arabidopsis ayant chacune une probabilité de germination p=0.8 donc selon B(100,0.8)
La probabilité que k=80 graines germent (k=nombre de succès)
dbinom(80,100,prob=0.8)
La probabilité que au maximum 80 graines germent
pbinom(80,100,prob=0.8)
La probabilité que plus de 80 graines germent
pbinom(80,100,prob=0.8, lower.tail=F)
En moyenne, combien de graines devraient germer ?
La distribution Binomiale B(100,0.8):
k=seq(0,100,1) # k=nombre de graines qui germent plot(dbinom(k,100,prob=0.8),xlab="k",ylab="Prob(X=k)")
Ajoutez ces parties à votre compte rendu.
Estimer un intervalle de confiance (IC) d'un paramètre populationnel (µ, p,...) à partir d'un échantillon
Intervalle de confiance de la moyenne théorique µ de la population, à partir de la moyenne m calculée sur un échantillon
Calculer l'intervalle de confiance (IC) du poids moyen théorique d'une récolte de tomates cerises, à partir d'un échantillon. Téléchargez et sauvegardez le fichier le fichier tomates_cerises.txt (click droit de la souris -- enregistrer la cible sous...).
Chargez le fichier avec la commande read.table dans une variable tomates, puis utilisez la commande attach pour accéder directement aux noms des colonnes. Utilisez également la commande names pour connaître le nom des colonnes dans le fichier. Affichez le contenu de la colonne poids_tomate.
Script pour calculer l'IC:
m = mean(poids_tomate) # moyenne calculée sur l'échantillon s = sd(poids_tomate) # écart-type calculé sur l'échantillon n = length(poids_tomate)# taille de l'échantillon
Si n≤100, on utilise la distribution de Student pour calculer la marge d'erreur. Ici, nous allons calculer l'intervalle ayant 95% de chance de contenir la vraie moyenne de la population (et pas celle de l'échantillon).
error <- qt(0.975,df=n-1)*s/sqrt(n) # qt(0.975) = quantile 97,5% de la distribution de Student ; s/sqrt(n) = erreur standard de la moyenne left <- m-error right <- m+error
L'intervalle ayant 95% de chance de contenir la vraie moyenne µ de la population est:
left;rightSi n>100 on utilise la distribution normale pour calculer l'erreur.
error <- qnorm(0.975)*s/sqrt(n) # qnorm(0.975) = quantile 97,5% de la distribution normale left <- m-error right <- m+error
L'intervalle ayant 95% de chance de contenir la vraie moyenne µ de la population est alors:
left;rightREMARQUE IMPORTANTE : si l'on veut un IC à 99% ==> remplacer 0.975 par 0.995.
IC de la proportion théorique p d'un caractère dans la population, à partir de la fréquence f calculée sur un échantillon
Calcul de l'IC de la proportion théorique de pommes rouges dans une récolte comportant des pommes rouges et vertes, à partir d'un échantillon de n=125 pommes contenant f=0.4 de pommes rouges (40%). On utilise une approximation normale de la distribution d'une proportion :
f <- 0.4 # fréquence de pommes rouges dans l'échantillon n <- 125 # taille de l'échantillon error <- qnorm(0.975)*sqrt(f*(1-f)/n) left <- f-error right <- f+error
L'intervalle ayant 95% de chance de contenir la vraie proportion p de la population est:
left;rightFaites varier la taille de l'échantillon ainsi que la confiance.
Ajoutez tout cela au compte rendu de TP avant de l'envoyer à votre enseignant par mail (maxime.bonhomme@univ-tlse3.fr ou roland.barriot@univ-tlse3.fr). Le compte rendu est à envoyer avant de commencer le TP3. Envoyez les 2 fichiers (.Rmd et .html). Envoyez-vous aussi le mail en copie pour pouvoir vérifier que tout est bien passé. Mettez un titre tel que "Compte rendu TP2 TDB de -et votre Nom et Prénom-". Votre compte-rendu ne doit pas être un simple "copié-collé" des commandes. Nous attendons une mise en forme correcte (titres, sous-titres, paragraphes, ...), des commentaires (notamment sur les résultats des commandes), des essais de votre part pour améliorer par exemple la visualisation d'un graphique, ou pour rechercher des fonctions R alternatives ou complémentaires à ce qui est proposé; bref une personnalisation de votre compte-rendu !
Liens
- Site de R : http://www.r-project.org et sites miroirs (dont ceux en France) pour télécharger le logiciel et les librairies : https://cran.r-project.org/mirrors.html
- RStudio : https://www.rstudio.com
- Utilisation de R depuis un navigateur : http://www.r-fiddle.org
Chargement des données avec l'adresse des fichiers
genomes=read.table("http://silico.biotoul.fr/site/images/d/de/Bacterial_genomes.txt", sep="\t", header=TRUE) sequoia=read.table("http://silico.biotoul.fr/site/images/e/e1/Sequoia.txt", sep="\t", header=TRUE) tomates=read.table("http://silico.biotoul.fr/site/images/3/3d/Tomates_cerises.txt", sep="\t", header=TRUE)