


TD3 Bioanalyse21
From silico.biotoul.fr
Contents |
OBJECTIFS
- Comprendre le résultat du programme BLAST - Utiliser un programme d'alignement multiple, et identifier des zones conservées, générer un Logo - Ecrire une signature protéique (pattern) - Rechercher dans une banque protéique des séquences qui possèdent cette signature
Exercice 1 : recherche de protéines homologues à BCL2 humaine
1/ Récupérer la protéine codée par l'ARNm humain NM_000633 (site du NCBI), et faites un InterProScan à l'EMBL-EBI (Services >InterProScan > copier coller votre séquence protéique dans le cadre > Search) pour caractériser l'organisation en domaines de cette protéine
- identifiez-vous des domaines dans les banques SMART (numero accession commençant par SMxx) et PROSITE (numero commençant par PSxxx)
- gardez cette page ouverte, ou notez la position des domaines
2/ Faites une analyse de type BlastP (proteine/proteine) contre la banque Uniprot/SwissProt au NCBI (Colonne de droite > BLAST > Blast Protein> Choisissez la banque Uniprot/SwissProt) pour identifier des séquences similaires à la protéine humaine
Regardez l'alignement avec la séquence BCL2-like protein 1 de poulet Q07816
- quelle est la taille de cette séquence ?
- à quoi correspond le % positives ?
- combien y a-t-il d'événement d'insertion-délétion ?
3/ Sélectionner (en cochant les cases à 'gauche' dans le tableau de l'onglet 'DESCRIPTIONS') les séquences qui vous semblent homologues à votre séquence 'QUERY' (prenez une vingtaine de séquences, a priori correspondant aux lignes rouges ou roses, de différents organismes)
4/ Récupérer les séquences en cliquant sur 'GenPept' en haut du tableau (puis Summary > FASTA text). Vous devez avoir une vingtaine de séquences au format FASTA
5/ Ouvrir/copier les séquences dans un editeur de texte (Word, Bloc note..) et renommer les pour avoir des noms courts : changer le nom dans l'entête FASTA, en gardant nom de protéine et de l'organisme, et sans espace (mais des tirets - ou _)
Vous pouvez simplifier le nom fourni dans SwissProt. Par exemple :
>sp|P10415.2|BCL2_HUMAN RecName: Full=Apoptosis regulator Bcl-2
devient :
>BCL2_HUMAN
Nb: éviter les espaces, parenthèses, préférez les _
Exercice 2 : Alignement multiple et construction d'une signature protéique
1/ Réaliser un alignement multiple sur le site de l'EBI utiliser MAFFT.
Pour cela : choisir dans le premier cadre 'Protein' > Copier_coller TOUTES vos sequences au format fasta dans le cadre du dessous > Choisir Output format : ClustalW> Cliquez sur Submit
- Regardez cet alignement et garder cette page ouverte !
2/ Visualiser dans une autre page l'alignement (onglet Result Viewers) avec Mview. Mview est juste une interface facilitant la visualisation de l'alignement (couleur)
- Où sont les parties conservées ? Notez les positions
- Voyez-vous apparaitre des groupes de séquences ?
3/ Copier/coller l'alignement (Onglet Result Summary au niveau de la 1ere page de l'alignement (avant Mview) => fichier tool output) dans WebLogo : modifier le paramètre Logo range pour cibler la zone conservée (voir selon votre alignement) et Logo Size per Line : 40 x 5 cm pour indiquer la taille de votre logo
4/ Construire une signature PROSITE à partir de votre alignement (n'utilisez pas le LOGO, sinon vous ne voyez pas précisément les zones de gap)
Pour vous aider, voici la début d'une signature (ou pattern) : Q-[IL]-H-x-T-[PQ]-x(2)-A-x(3)-F
Vous avez probablement quelque chose d'approchant dans votre alignement (vers 170), traduisant que :
Q : il n'y a que l' acide aminé Q dans cette colonne de l'alignement
[IL] : dans cette colonne seuls les acides aminés I ou L sont présents
x(2) : 2 colonnes avec des acides aminés variables
Vous pouvez visualiser des exemples de signature ici.
5/ Tester votre signature sur ScanProsite (choisir l'option 2) contre SwissProt (ou trEMBL analyse plus longue !)
- Les séquences obtenues appartiennent-elles à la famille des BCL2 ou BCL2-like ?
- Retrouvez-vous les mêmes organismes que précédemment ? en avez-vous d'autres ?
6/ Regarder, par exemple dans le lien UniProt Q07816 (B2CL1_CHICK), comment est caractérisé ce domaine dans les banques : dans la partie Family and domain databases cliquez sur View potein in PROSITE : est-ce-que ce sont plutôt des signatures ou des profiles ?
On retiendra que les signatures (pattern en anglais) sont plutôt utilisées pour les motifs (régions assez courtes et bien conservées) et les profiles (matrices PSSM) pour des domaines protéiques
BONUS pour ceux qui ont encore du temps et/ou qu'ils veulent faire des arbres !!:
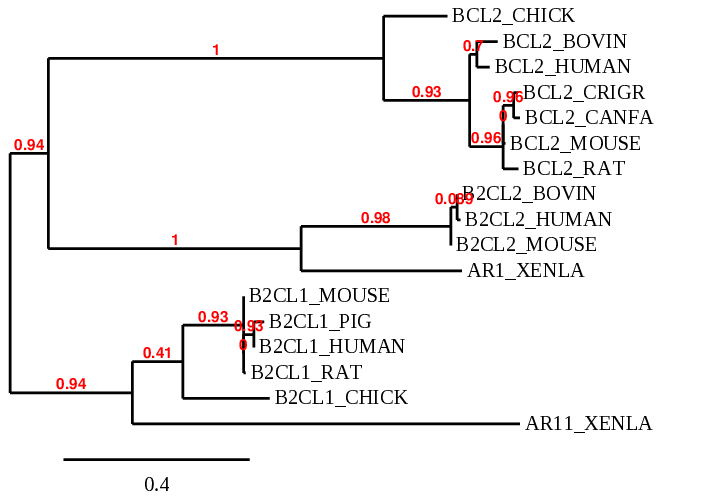
7/ Pour finir, vous pouvez générer un arbre phylogénétique de vos séquences : aller sur sur le site Phylogeny dans Phylogeny Analysis => "Advanced" : décocher la case Multiple alignment et celle Gblocks, et cliquer sur Create workflow. Coller votre alignement multiple. Vous pouvez aussi utiliser le mode "One click" et mettre directement les séquences non alignées en format Fasta (là aussi décocher Gblocks).
Essayer de comprendre l'histoire de cette famille et de voir les nœuds de spéciation et de duplication.
Annexes
Résultat InterProScan
Résultats du Blast P
Jeu de Séquences
ScanProsite contre TrEMBL
Arbre phylogénétique
{kind=link}